PROYECTO de Norma Oficial Mexicana PROY-NOM-127-SSA1-2017, Agua para uso y consumo humano. Límites permisibles de la calidad del agua.Al margen un sello con el Escudo Nacional, que dice: Estados Unidos Mexicanos.- Secretaría de Salud. JOSÉ ALONSO NOVELO BAEZA, Comisionado Federal para la Protección contra Riesgos Sanitarios y Presidente del Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario, con fundamento en lo dispuesto por los artículos 39 de la Ley Orgánica de la Administración Pública Federal; 4 de la Ley Federal de Procedimiento Administrativo; 3o., fracción XIII, 13, apartado A, fracción I, 17 bis, fracciones II y III, 116, 118, fracción II y 119, fracción II de la Ley General de Salud; 38, fracción II, 40, fracción I, 43 y 47, fracción I de la Ley Federal sobre Metrología y Normalización; 28 y 33 del Reglamento de la Ley Federal sobre Metrología y Normalización; 209 a 213, 214, fracciones I, II, III y V, 215 a 225 y 227 del Reglamento de la Ley General de Salud en Materia de Control Sanitario de Actividades, Establecimientos, Productos y Servicios; así como 3, fracciones I, incisos n), o) y s), y II, 10, fracción IV del Reglamento de la Comisión Federal para la Protección contra Riesgos Sanitarios; he tenido a bien ordenar la publicación en el Diario Oficial de la Federación el PROYECTO DE NORMA OFICIAL MEXICANA PROY-NOM-127-SSA1-2017, AGUA PARA USO Y CONSUMO HUMANO. LÍMITES PERMISIBLES DE LA CALIDAD DEL AGUA El presente Proyecto se publica a efecto de que los interesados, dentro de los 60 días naturales siguientes a la fecha de su publicación en el Diario Oficial de la Federación, presenten sus comentarios por escrito, en idioma español y con el sustento técnico correspondiente, ante el Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario, sito en Oklahoma número 14, planta baja, colonia Nápoles, Alcaldía Benito Juárez, Código Postal 03810, Ciudad de México, teléfono 50805200, extensión 1333, correo electrónico rfs@cofepris.gob.mx. Durante el plazo mencionado y de conformidad con lo dispuesto por los artículos 45 y 47, fracción I de la Ley Federal sobre Metrología y Normalización, los documentos que sirvieron de base para la elaboración del Proyecto y su Análisis de Impacto Regulatorio estarán a disposición del público en general, para su consulta, en el domicilio del mencionado Comité. PREFACIO En la elaboración de este Proyecto de Norma participaron: SECRETARÍA DE SALUD Comisión Federal para la Protección contra Riesgos Sanitarios SECRETARÍA DE MEDIO AMBIENTE Y RECURSOS NATURALES Comisión Nacional del Agua Instituto Mexicano de Tecnología del Agua SECRETARÍA DE ENERGÍA Comisión Nacional de Seguridad Nuclear y Salvaguardias SISTEMA DE AGUAS DE LA CIUDAD DE MÉXICO Subdirección de Control de la Calidad del Agua COMISIÓN DEL AGUA DEL ESTADO DE MÉXICO Departamento del Laboratorio del Agua SERVICIOS DE AGUA Y DRENAJE DE MONTERREY, I.P.D. Laboratorio Central de Calidad de Aguas ORGANIZACIÓN MUNDIAL DE LA SALUD Organización Panamericana de la Salud UNIVERSIDAD NACIONAL AUTÓNOMA DE MÉXICO Instituto de Ecología Laboratorio Nacional de Ciencias de la Sostenibilidad INSTITUTO POLITÉCNICO NACIONAL Escuela Nacional de Ciencias Biológicas UNIVERSIDAD AUTÓNOMA DE SAN LUIS POTOSÍ Centro de Investigación Aplicada en Ambiente y Salud Confederación de Cámaras Industriales de los Estados Unidos Mexicanos Confederación Patronal de la República Mexicana Cámara Nacional de la Industria de Transformación Asociación Nacional de Empresas de Agua y Saneamiento de México, A.C. Laboratorios ABC Química Investigación y Análisis, S.A. de C.V. Laboratorios de Especialidades Inmunológicas, S.A. de C.V. ÍNDICE 0. Introducción 1. Objetivo y campo de aplicación 2. Referencias normativas 3. Términos y definiciones 4. Símbolos y términos abreviados 5. Especificaciones sanitarias 6. Métodos de prueba 7. Concordancia con normas internacionales 8. Procedimiento de evaluación de la conformidad 9. Bibliografía 10. Observancia de la Norma 11. Vigencia Apéndice A Normativo. Parámetros que conforman los grupos de compuestos orgánicos sintéticos Apéndice B Normativo. Métodos de prueba Apéndice C Informativo. Procesos propuestos para la potabilización del agua 0. Introducción El abastecimiento de agua para uso y consumo humano con calidad adecuada es fundamental para prevenir y evitar la transmisión de enfermedades relacionadas con el agua, para lo cual se requiere establecer y mantener actualizados los límites permisibles en cuanto a sus características físicas, químicas, microbiológicas, y radiactivas, con el fin de asegurar y preservar la calidad del agua que se entrega al consumidor por los sistemas de abastecimiento de agua públicos y privados. Por tales razones la Secretaría de Salud, propone la emisión de la presente Norma Oficial Mexicana, con la finalidad de establecer un eficaz control sanitario del agua que se somete a tratamientos de potabilización a efecto de hacerla apta para uso y consumo humano, acorde a las necesidades actuales. 1. Objetivo y campo de aplicación 1.1 Esta Norma establece los límites permisibles de calidad que debe cumplir el agua para uso y consumo humano. 1.2 Esta Norma es de observancia obligatoria en el territorio nacional para los organismos responsables de los sistemas de abastecimiento de agua públicos y privados. 1.3 Esta Norma no es aplicable para aguas residuales tratadas. 2. Referencias normativas Para la correcta aplicación de esta Norma, es necesario consultar las siguientes normas oficiales mexicanas o las que las sustituyan: 2.1 Norma Oficial Mexicana NOM-008-SCFI-2002, Sistema General de Unidades de Medida. 2.2 Norma Oficial Mexicana NOM-117-SSA1-1994, Bienes y servicios. Método de prueba para la determinación de cadmio, arsénico, plomo, estaño, cobre, fierro, zinc y mercurio en alimentos, agua potable y agua purificada por espectrometría de absorción atómica. 2.3 Norma Oficial Mexicana NOM-201-SSA1-2015, Productos y servicios. Agua y hielo para consumo humano, envasados y a granel. Especificaciones sanitarias. 2.4 Norma Oficial Mexicana NOM-210-SSA1-2014, Productos y servicios. Métodos de prueba microbiológicos. Determinación de microorganismos indicadores. Determinación de microorganismos patógenos. 3. Términos y definiciones Para los propósitos de esta Norma, se aplican los términos y definiciones siguientes: 3.1 Agua para uso y consumo humano, a toda aquella que no causa efectos nocivos a la salud y que no presenta propiedades objetables o contaminantes en concentraciones fuera de los límites permisibles y que no proviene de aguas residuales tratadas. 3.2 Aguas residuales, a las de composición variada provenientes de las descargas de usos público, urbano, doméstico, industrial, comercial, de servicios, agrícola, pecuario, de las plantas de tratamiento y en general, de cualquier uso, así como la mezcla de ellas. 3.3 Agua superficial, a la que fluye sobre la superficie del suelo a través de arroyos, canales y ríos, o que se almacene en lagos, embalses, ya sean naturales o artificiales. 3.4 Brote, a la ocurrencia de dos o más casos asociados epidemiológicamente entre sí. La existencia de un caso único bajo vigilancia especial en un área donde no existía el padecimiento se considera también como brote. 3.5 Compuestos orgánicos halogenados adsorbibles fijos, al grupo de parámetros analíticos que comprende compuestos orgánicos con halógenos que no son volátiles. 3.6 Compuestos orgánicos halogenados adsorbibles purgables, al grupo de parámetros analíticos que comprende compuestos orgánicos con halógenos que son volátiles. 3.7 Compuestos orgánicos no halogenados, al grupo de parámetros analíticos que comprende compuestos orgánicos semivolátiles sin halógenos. 3.8 Desinfección, al proceso físico y/o químico utilizado para la eliminación, inactivación o destrucción de microorganismos patógenos en el agua. 3.9 Emergencia, a cualquier incidente o accidente en los componentes del sistema de abastecimiento de agua, que dé lugar a alteraciones en la calidad del agua que represente un riesgo a la salud de la población. 3.10 Límite permisible, al valor máximo o intervalo de valores establecidos para los parámetros físicos, químicos, microbiológicos o radiactivos, que no deben excederse en el agua para uso y consumo humano. 3.11 Método de prueba, al procedimiento analítico utilizado en el laboratorio para comprobar que el agua satisface las especificaciones de esta Norma. 3.12 Organismo responsable, a la instancia encargada de operar, mantener y administrar el sistema de abastecimiento de agua con el fin de asegurar y preservar la calidad del agua que se entrega al consumidor por los sistemas de abastecimiento de agua públicos y privados, estableciendo un eficaz control sanitario del agua sometiéndola a tratamientos de potabilización a efecto de hacerla y mantenerla apta para uso y consumo humano. 3.13 Potabilización, al conjunto de operaciones y procesos, físicos, químicos y biológicos que se aplican al agua en los sistemas de abastecimiento de agua, a fin de hacerla apta para uso y consumo humano. 3.14 Sistema de abastecimiento de agua, al conjunto intercomunicado o interconectado de fuentes, obras de captación, plantas potabilizadoras, tanques de almacenamiento y regulación, líneas de conducción y distribución, incluyendo vehículo cisterna que abastece de agua para uso y consumo humano, sean de propiedad pública o privada. 4. Símbolos y términos abreviados Cuando en esta Norma se haga referencia a los siguientes símbolos y abreviaturas, se entiende por: 4.1 Bq becquerel 4.2 Bq/L becquerel por Litro 4.3 CaCO3 carbonato de calcio 4.4 DDT diclorodifeniltricloroetano 4.5 Di prefijo en nomenclatura química que indica que el sustituyente se encuentra dos veces 4.6 E. coli Escherichia coli 4.7 F- ión fluoruro 4.8 L litro 4.9 LR levo-rotativo 4.10 mg miligramo 4.11 mL mililitro 4.12 µg microgramo 4.13 NMP número más probable 4.14 N-NO3- nitrógeno de nitratos 4.15 N-NO2- nitrógeno de nitritos 4.16 pH potencial de hidrógeno 4.17 SO4= sulfatos 4.18 UC unidades de color 4.19 UFC unidades formadoras de colonias 4.20 UNT unidades nefelométricas de turbiedad 4.21 2,4-D ácido 2,4-diclorofenoxiacético 4.22 2,4-DB ácido 2,4-diclorofenoxibutírico 4.23 2,4,5-T ácido 2,4,5 triclorofenoxiacético 4.24 2,4,5-TP ácido 2,4,5-triclorofenoxi propiónico 4.25 < menor que 5. Especificaciones sanitarias El agua para uso y consumo humano de los sistemas de abastecimiento debe cumplir con las siguientes especificaciones: 5.1 El agua de los sistemas de abastecimiento no debe tener como fuente de abastecimiento agua residual tratada. 5.2 Físicas: Tabla 1 - Especificaciones sanitarias físicas
5.3 Químicas: Tabla 2 - Especificaciones sanitarias químicas
Tabla 3 - Tabla de cumplimiento gradual para fluoruro
5.4 Metales y metaloides: Tabla 4 - Especificaciones sanitarias de metales y metaloides
Tabla 5 - Tabla de cumplimiento gradual para arsénico y cadmio
5.5 Microbiológicas: Tabla 6 - Especificaciones sanitarias microbiológicas
5.6 Fitotoxinas: Tabla 7 - Especificaciones sanitarias de fitotoxinas
5.7 Radiactividad: Tabla 8 - Especificaciones sanitarias de radiactividad
5.8 Residuales de la desinfección: 5.8.1 Si para la desinfección del agua se utiliza algún compuesto de cloro (hipoclorito de sodio o de calcio, gas cloro o dióxido de cloro) debe medirse cloro residual libre. 5.8.2 Si para la desinfección del agua se utiliza yodo debe medirse yodo residual libre. 5.8.3 Si para la desinfección del agua se utiliza cualquier forma de plata debe medirse plata total. Tabla 9 - Especificaciones sanitarias de residuales de la desinfección
5.8.4 La Secretaría de Salud determinará si un agente diferente a los establecidos en la Tabla 9 puede ser utilizado para la desinfección de agua para uso y consumo humano. 5.8.5 En caso de un brote, para garantizar la protección a la población, la Secretaría de Salud determinará el agente desinfectante del agua y el intervalo de concentración de los residuales de desinfección. 5.9 Subproductos de la desinfección. 5.9.1 Si el agua se desinfecta con algún compuesto de cloro se deben medir los subproductos de la desinfección: trihalometanos y ácidos haloacéticos, de conformidad con lo establecido en las Tablas 10 y 11 de esta Norma. Tabla 10 - Especificaciones sanitarias de subproductos de la desinfección - trihalometanos
Tabla 11 - Especificaciones sanitarias de subproductos de la desinfección - ácidos haloacéticos
5.9.2 Si el agua se desinfecta con ozono, se deben medir los subproductos de la desinfección: aniones y carbonilos, de conformidad con lo establecido en las Tablas 12 y 13 de esta Norma. Tabla 12 - Especificaciones sanitarias de subproductos de la desinfección - aniones
Tabla 13 - Especificaciones sanitarias de subproductos de la desinfección - carbonilos
5.10 Compuestos orgánicos sintéticos: Tabla 14 - Especificaciones sanitarias de compuestos orgánicos sintéticos
5.10.1 En caso de sobrepasar alguno de los límites permisibles de los grupos de compuestos orgánicos sintéticos de la Tabla 14 de esta Norma, el organismo responsable deberá analizar los compuestos orgánicos asociados establecidos en el Apéndice A Normativo de esta Norma correspondientes al grupo de compuestos orgánicos sintéticos que sobrepase el límite permisible. 5.11 Cuando se excedan los límites permisibles expuestos en este Capítulo, se deben aplicar los procesos de tratamiento adecuados para su remoción, entre los cuales puede aplicar el que corresponda, de los referidos en el Apéndice C Informativo de esta Norma o cualquier otro que sea efectivo para la remoción del contaminante. 5.12 En caso de alguna emergencia (ver inciso 3.9 de esta Norma), la Secretaría de Salud definirá los parámetros y límites aplicables al agua destinada para uso y consumo humano, conforme a lo dispuesto por el artículo 116 de la Ley General de Salud. 6. Métodos de prueba 6.1 Para la determinación de las especificaciones fisicoquímicas de agua, se deben aplicar los métodos que establecen las normas oficiales mexicanas citadas en los puntos 2.2 y 2.3 del Capítulo de Referencias normativas o los establecidos en el Apéndice B Normativo, Métodos de prueba de esta Norma. 6.2 Para la determinación de las especificaciones microbiológicas de agua, se deben aplicar los métodos de prueba que establece la norma oficial mexicana citada en el punto 2.4 del Capítulo de Referencias normativas o los establecidos en el Apéndice B Normativo Métodos de prueba de esta Norma. 7. Concordancia con normas internacionales Esta Norma no es concordante con ninguna Norma Internacional. 8. Procedimiento de evaluación de la conformidad La evaluación de la conformidad podrá ser solicitada a instancia de parte por el responsable sanitario, el representante legal o la persona que tenga las facultades para ello, ante la autoridad competente o las personas acreditadas y autorizadas para tales efectos. 9. Bibliografía 9.1 American Public Health Association (2012). Standard Methods for the Examination of Water and Wastewater, 22nd Edition. Washington D.C. EUA. 9.2 American Water Works Association (2000). Water Quality and Treatment: A Handbook of Community Water Supplies, 5th Edition, McGraw Hill Inc, USA. 9.3 Betancourt-Lineares, A., Irigoyen-Camacho, ME., Mejía-González, A., Zepeda-Zepeda, M. & Sánchez-Pérez, L. (2013). Prevalencia de Fluorósis Dental en Localidades Mexicanas Ubicadas en 27 Estados y el DF. A Seis Años de la Publicación de la Norma Oficial Mexicana para la Fluoruración de la Sal. Revista de Investigación Clínica. 65, (3) 23, pp 7-24. 9.4 Bocanegra-Salazar, M., Landín-Rodríguez, LE. & Ortíz-Pérez, MD. (2006). Tesis: Evaluación de la Contaminación por Flúor y Arsénico en el Agua de Pozo para Consumo Humano de las Zonas Centro, Altiplano y media del Estado de San Luis Potosí. Universidad Autónoma de San Luis Potosí. 9.5 Comisión Nacional del Agua (2015). Manual de Agua Potable, Alcantarillado y Saneamiento (Mapas). 9.6 Programa de las Naciones Unidas para el Medio Ambiente. 2001. Convenio de Estocolmo sobre Contaminantes Orgánicos Persistentes. UNEP. 9.7 Das K, Mondal NK. (2016). Dental fluorosis and urinary fluoride concentration as a reflection of fluoride exposure and its impact on IQ level and BMI of children of Laxmisagar, Simlapal Block of Bankura District, W.B., India. Environmental Monitoring & Assessment 188(4):218. 9.8 Environmental Protection Agency (2010). Guidelines for Design of Small Public Ground Water Systems. Fourth Edition. Division of drinking and groundwater. Ohio. p76. 9.9 Hurtado-Jiménez, R. & Gardea-Torresdey, JL. (2006). Arsenic in Drinking Water in the Los Altos de Jalisco Region of Mexico. Rev. Panam. Salud Pública. 20, (4): pp 23647. 9.10 Khan SA, et al. (2015). Relationship between dental fluorosis and intelligence quotient of school going children in and around Lucknow district: a cross-sectional study. Journal of Clinical & Diagnostic Research 9(11):ZC10-15. 9.11 Ley de Aguas Nacionales. 9.12 Loyola-Rodríguez, JP., Pozos-Guillén AJ., Hernández-Guerrero, JC. & Hernández-Sierra JF. (2000). Fluorosis en Dentición Temporal en un Área con Hidrofluorosis Endémica. Salud Pública de México. 42, pp 194-200. 9.13 Mondal D, et al. (2016). Inferring the fluoride hydrogeochemistry and effect of consuming fluoride-contaminated drinking water on human health in some endemic areas of Birbhum district, West Bengal. Environmental Geochemistry & Health 38(2):557-76. 9.14 Norma Oficial Mexicana NOM-017-SSA2-2012. Para la Vigilancia Epidemiológica. http://dof.gob.mx/nota_detalle.php?codigo=5288225&fecha=19/02/2013 9.15 Panduro Rivera, MG. (2015). Evaluación de la Calidad del Agua Ante la Enfermedad Renal Crónica en la Zona Oriente de Michoacán. Tesis Maestro de Maestría. Centro de Investigación y Asistencia en Tecnología y Diseño del Estado de Jalisco, AC. 9.16 Sebastian ST, Sunitha S. 2015. A cross-sectional study to assess the intelligence quotient (IQ) of school going children aged 10-12 years in villages of Mysore district, India with different fluoride levels. Journal of the Indian Society of Pedodontics and Preventive Dentistry 33(4):307-11. 9.17 Secretaría de Salud (2012). Manual de Procedimientos Estandarizados para la Vigilancia Epidemiológica de las Patologías Bucales. http://www.cenaprece.salud.gob.mx/programas/interior/saludbucal/descargas/pdf/20_2012_Manual_PatBucales_vFinal.pdf 9.18 World Health Organization (2017). Guidelines for Drinking-Water Quality. 4a edición incorporando el primer addendum. 9.19 World Health Organization (2010). Diez sustancias químicas que constiutyen una preocupación para la salud pública. http://www.who.int/ipcs/assessment/public_health/chemicals_phc/es/ 10. Observancia de la Norma La vigilancia del cumplimiento de esta Norma corresponde a la Secretaría de Salud a través de la Comisión Federal para la Protección contra Riesgos Sanitarios y a los gobiernos de las entidades federativas, en sus respectivos ámbitos de competencia. 11. Vigencia Esta Norma entrará en vigor a los 360 días naturales contados a partir del día siguiente de su publicación en el Diario Oficial de la Federación. TRANSITORIO ÚNICO.- La entrada en vigor de la presente Norma, deja sin efectos a la Modificación a la Norma Oficial Mexicana NOM-127-SSA1-1994, Salud Ambiental. Agua para uso y consumo humano. Límites permisibles de calidad y tratamientos a que debe someterse el agua para su potabilización, publicada en el Diario Oficial de la Federación el 22 de noviembre de 2000. Ciudad de México, a 10 de octubre de 2019.- El Comisionado Federal para la Protección contra Riesgos Sanitarios y Presidente del Comité Consultivo Nacional de Normalización de Regulación y Fomento Sanitario, José Alonso Novelo Baeza.- Rúbrica. APÉNDICE A NORMATIVO Parámetros que conforman los grupos de compuestos orgánicos sintéticos Tabla A.1 Límites permisibles de compuestos orgánicos halogenados adsorbibles fijos
Tabla A.2 Límites permisibles de compuestos orgánicos no halogenados
Tabla A.3 Límites permisibles de compuestos orgánicos halogenados adsorbibles purgables
APÉNDICE B NORMATIVO Métodos de prueba B.1 MÉTODOS PARA LA DETERMINACIÓN DE MICROCISTINA EN AGUA PARA USO Y CONSUMO HUMANO Para el cumplimiento de esta Norma, se deberá utilizar el método de ELISA cualitativo de respuesta rápida (screening) para la determinación de microcistinas y nodularinas totales, contenido en el punto B.1.1 de este Apéndice). En caso de que dicho método resulte en la ausencia de microcistinas o nodularinas totales no será necesario llevar a cabo ningún método confirmatorio adicional. En caso de que dicho método resulte en la presencia de microcistinas o nodularinas totales deberá de llevarse a cabo alguno de los dos métodos cuantitativos confirmatorios siguientes: · Método para la determinación de microcistinas y nodularinas en agua de uso y consumo humano por extracción en fase sólida y cromatografía líquida/espectrometría de masas en tándem (LC/MS/MS), contenido en el punto B.1.2 de este Apéndice. · Método para la determinación de microcistina mediante SPE y la cromatografía de líquidos de alto rendimiento (HPLC) con detección (UV) ultravioleta, contenido en el punto B.1.3 de este Apéndice. Sin embargo, para la determinación de microcistina-LR podrá optarse por determinar directamente dicha sustancia a través de los métodos confirmatorios cuantitativos descritos en los puntos B.1.2 y B.1.3 de este Apéndice, sin llevar a cabo el método de screening ELISA (B.1.1, de este Apéndice). B.1.1 MÉTODO PARA LA DETERMINACIÓN DE MICROCISTINAS Y NODULARINAS TOTALES EN AGUA DE USO Y CONSUMO HUMANO MEDIANTE EL ENSAYO INMUNOENZIMÁTICO ADDA B.1.1.1 Definiciones y términos Agua grado reactivo, al agua purificada que no contiene ninguna cantidad cuantificable de microcistinas, nodularinas o compuestos que interfieran en o por encima de la media del MRL. Blanco de reactivo de laboratorio (LRB), a la alícuota de agua grado reactivo lisada y filtrada para coincidir en el mismo procedimiento analítico utilizado para las muestras de campo. El LRB se utiliza para determinar si se introducen microcistinas u otras interferencias por medio de los recipientes de la muestra, los equipos de procesamiento de muestras o los reactivos utilizados en el ensayo. Blanco fortificado de laboratorio (LFB), a la alícuota de agua grado reactivo a la que se añade una cantidad conocida de MC-LR. El LFB es lisado y filtrado para coincidir en el mismo procedimiento analítico utilizado para las muestras de campo. El LFB se utiliza durante la IDC para verificar el rendimiento del método con precisión y exactitud. El LFB también es un elemento de control de calidad requerido con cada lote de análisis. Los resultados del LFB verifican el rendimiento del método en ausencia de matriz de muestra. Concentración más baja del nivel mínimo reportado (LCMRL), al punto de concentración más baja, tal que la probabilidad de recobro se encuentre entre el 50 y 150% con al menos 99% de confianza. Curva de calibración, a los puntos de calibración se modelan utilizando una función logística de cuatro parámetros, relacionando la concentración (eje x) con la absorbancia medida en los pozos (eje y). A continuación se presenta un ejemplo de curva de calibración generada durante el desarrollo del método. Es importante observar la relación inversa entre la concentración y la respuesta (absorbancia). 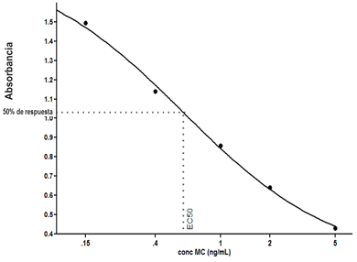 Fig. B.1.1-1. Curva de calibración de ELISA con cálculo de la EC50. El eje x se encuentra en escala logarítmica. El estándar de calibración cero proporciona la absorbancia más alta y el estándar de calibración más alto proporciona la absorbancia más baja. De igual manera, es importante observar también que la pendiente, o sensibilidad, de la respuesta ELISA es mayor en el centro de la curva y tiende a una pendiente de cero a concentraciones extremadamente bajas y altas. Para una explicación más detallada del modelo de calibración de cuatro parámetros pueden revisarse referencias específicas como Maciel (1985) y Sasaki (2016). Ecuación logística de cuatro parámetros: 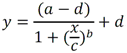 En donde: y absorbancia x concentración a absorbancia en la meseta inferior b pendiente en el punto de inflexión c concentración en el punto de inflexión = EC50 d absorbancia en la meseta superior Los coeficientes a, b, c y d, son calculados por el software de reducción de datos usando análisis de regresión. Duplicado de la matriz de muestra fortificada en laboratorio (LFSMD), a la segunda alícuota de la muestra de campo utilizada para preparar el LFSM que se fortifica y ensaya en el mismo lote de análisis que el LFSM. El LFSMD se utiliza para verificar la precisión del método en las matrices de agua de uso y consumo humano. EC50, a la concentración de microcistina que produce una absorbancia media entre la meseta inferior de la curva de calibración (coeficiente a) y la meseta superior (coeficiente d). La EC50 es la concentración en el punto de inflexión (Fig. B.1.1-1 de este Apéndice B) y está en el centro del intervalo de medición más confiable (es decir, la mayor pendiente) de la ELISA. Para cada curva de calibración, la EC50 es igual al coeficiente c, del ajuste logístico de cuatro parámetros. El EC50 se encuentra en el informe de calibración generado por el lector de placas. Debido a que la EC50 está en el centro del intervalo de medición más confiable, la guía para fortificar muestras de control de calidad se basa en este valor. Para este método, las muestras de control de calidad que requieren ser fortificadas con MC-LR deben tener concentraciones cerca de la EC50. Estas muestras de control de calidad incluyen blancos fortificados de laboratorio y matriz de muestras fortificadas de laboratorio. Estándar de calibración, soluciones de MC-LR suministradas en el kit de la prueba de ELISA o preparadas en el laboratorio, que son apropiadas para el rango de medición del kit de ELISA. Solución estándar de dilución primaria (PDS), solución de MC-LR en metanol (o el indicado por el fabricante) preparado a partir de la solución estándar MC-LR. Las soluciones de PDS se utilizan para fortificar muestras de control de calidad (LFB, LFSM y LFSMD). Estándar de verificación de calibración de bajo alcance (LOW-CV), estándar de calibración con una concentración igual o inferior al MRL. El propósito de la LOW-CV es confirmar la exactitud de la calibración en concentraciones cercanas al MRL. Lote de análisis, los estándares, muestras y elementos de control de calidad se ensayan en una sola placa de 96 pozos usando lotes idénticos de reactivos y pozos. Cada placa por definición es un lote de análisis, independientemente del número de pozos incluidos. Las muestras de control de calidad deben analizarse en cada lote de análisis a las frecuencias descritas. Cada lote de análisis incluye los siguientes elementos: Estándares de calibración Blancos de reactivo de laboratorio Estándar de verificación de calibración de bajo alcance Blancos fortificados de laboratorio Muestras de campo (agua de uso y consumo humano) Matriz de muestras fortificadas en laboratorio y duplicados de las matrices de muestras fortificadas en laboratorio. Matriz de muestra fortificada en laboratorio (LFSM), alícuota de una muestra de campo a la que se añade una cantidad conocida de MC-LR. El propósito de la LFSM es determinar si la matriz de la muestra contribuye a los resultados analíticos. Muestra de control de calidad (QCS), solución que contiene MC-LR en una concentración conocida que se obtiene de una fuente diferente de la fuente de los estándares de calibración. El propósito de la QCS es verificar la exactitud de los estándares de calibración primaria. Nivel mínimo reportado (MRL), concentración mínima que puede ser reportada por un laboratorio como un valor cuantificado para microcistinas y nodularinas totales en una muestra después del análisis. Esta concentración no debe ser inferior a la concentración del estándar de calibración más bajo y debe cumplir con los criterios de control de calidad. Pozos replica, dentro del lote de análisis, este método requiere que cada estándar de calibración, muestra de campo y muestra de control de calidad se ensayen en dos pozos. Estos dos pozos se denominan pozos replica. A cada pozo replica se asocian dos valores: la absorbancia medida por el lector de placas y la concentración calculada a partir de esta absorbancia. Para cada conjunto de pozos replica, el coeficiente de variación porcentual (% CV) se calcula a partir de los dos valores de absorbancia. El %CV de los valores de absorbancia para los estándares de calibración debe cumplir con los criterios de control de calidad especificados. El %CV de los valores de absorbancia para todas las muestras de campo y las muestras de control de calidad deben cumplir con los criterios de control de calidad. Para cada conjunto de pozos replica, se calcula la concentración media de los dos valores de concentración. La concentración media debe usarse para reportar los resultados de la muestra de campo. La media debe utilizarse en todos los cálculos del método y para evaluar los resultados con respecto a los límites de control de calidad. Solución estándar, estándar concentrado en metanol (o el indicado por el fabricante) que se prepara en el laboratorio a partir de MC-LR purificada o que se adquiere de una fuente comercial con un certificado de análisis. B.1.1.2 Símbolos y términos abreviados (4E, 6E) posición espacial de los alquenos en la molécula °C grado Celsius DPD N, N-dietil-p-fenilendiamina IDC demostración inicial de capacidad LCMRL concentración más baja del nivel mínimo reportado LFB blanco fortificado de laboratorio LFSM matriz de muestra fortificada en laboratorio LFSMD duplicado de la matriz de muestra fortificada en laboratorio LOW-CV estándar de verificación de calibración de bajo alcance LRB blanco de reactivo de laboratorio MC microcistina MC/NOD relación microcistina entre nodularina MC-LR microcistina-LR mm milímetro MRL nivel mínimo reportado µg/ml microgramo/mililitro µm micrómetro µL microlitro nm nanómetro NOD nodularina NOD-R Nodularina-R PDS solución estándar de dilución primaria PIR intervalo de predicción de resultados PTFE politetrafluoroetileno QCS muestra de control de calidad RPD diferencia porcentual relativa r2 cuadrado del coeficiente de correlación % por ciento %CV coeficiente de variación porcentual %R porcentaje promedio de recuperación %RSD porcentaje de desviación estándar relativa menor o igual a > mayor a mayor o igual a B.1.1.3 Principio El método descrito es un procedimiento mediante ensayo inmunoenzimático (ELISA). Este método se basa en un sistema de placas de microtitulación de 96 pozos, en estos pozos, las microcistinas y nodularinas presentes en la muestra y proteína análoga de microcistina inmovilizada en los pozos, compiten por los sitios de unión de un anticuerpo de detección primaria en solución. Después de una etapa de lavado, se añade un conjugado enzimático a los pozos y se une al anticuerpo primario en una relación inversa con la concentración original de microcistinas y nodularinas en la muestra. Después de un segundo paso de lavado, se añade substrato de tetrametilbenzidina para desarrollar color a través de una reacción enzimática. Tras un período establecido, se añade una solución ácida a cada pozo para detener la generación de color. Finalmente, la absorbancia de cada pozo se mide utilizando un lector de placas y la concentración de microcistinas y nodularinas se calcula utilizando una curva de calibración logística de cuatro parámetros. B.1.1.4 Alcance y aplicación El método descrito es un procedimiento para la determinación de microcistinas (MC) y nodularinas (NOD) "totales" en agua de uso y consumo humano (antes y después del tratamiento). El término "microcistinas y nodularinas totales" se define como la suma de todos los congéneres independientes, tanto de microcistinas intracelulares y extracelulares, como de nodularinas que es medible en una muestra. Este método determina la concentración total basada en la detección de una característica común a los congéneres (variantes estructurales) de microcistina y nodularina, específicamente la cadena lateral de aminoácidos Adda: Ácido (4E, 6E)-3-amino-9-metoxi-2,6,8-trimetil-10-fenildeca-4,6-dienoico (Fischer et al., 2001). Con la finalidad de asegurar la comparabilidad entre los laboratorios, el ELISA se calibra contra el congénere Microcistina-LR (MC-LR) (CASRN 101043-37-2). B.1.1.5 Equipos y materiales Las referencias a marcas específicas y números de catálogo se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden representar especificaciones adecuadas para los artículos. Contenedores de muestras. Botellas de vidrio ámbar con tapas con rosca de politetrafluoroetileno (PTFE). Se recomienda el uso de contenedores de muestras. Si se reutilizan contenedores de muestra deben de llevarse a cabo buenas prácticas de laboratorio para la limpieza. No deben utilizarse botellas tratadas a alta temperatura en un horno de mufla (400 °C y superiores) como procedimiento de limpieza. Los estudios existentes indican una tendencia a que las microcistinas y las nodularinas se adsorben en la superficie de la cristalería limpiada repetidamente por calentamiento en un horno de mufla. Cubiertas de placas adhesivas. Película adhesiva transparente para sellar los pozos durante las etapas de incubación del ensayo (VWR no. 60941-120). Filtros de jeringa para filtrado de muestras después de la lisis. Pueden utilizarse filtros de fibra de vidrio de 25mm, con un tamaño de poro de 1.2µm y carcasa de polipropileno (Environmental Express no. SF012G) o filtros de fibra de vidrio de 25 mm, con un tamaño de poro de 0.45µm y carcasa de polipropileno (GE Healthcare Life Sciences/Whatman no. 6894-2504). Jeringas para filtrado de muestras después de la lisis. Pueden utilizarse jeringas de cristal con cerrado Luer-lock que no permita el paso de aire, de 5mL de capacidad (Hamilton Co. no. 1005TLL) o jeringas de plástico con cerrado Luer-lock que no permita el paso de aire, con una capacidad de 3 mL y cilindros de polietileno y émbolos de polipropileno (Thermo Fisher Scientific, Inc. no. S7515-3). Kit de prueba de ELISA Adda. Ensayo competitivo indirecto basado en la detección del epítopo Adda (Abraxis no. 520011OH o equivalente). Pueden usarse sistemas automatizados para procesar la placa de 96 pozos. Lector de Placas. Lector de placas de microtitulación y software asociado con la capacidad de determinar la absorbancia a 450 nm y construir una curva de calibración utilizando una función logística de cuatro parámetros. Pipeta de repetición. Pipeta electrónica de repetición HandyStep con puntas desechables de 5 ml (Wertheim, Alemania). Se recomienda una pipeta electrónica de repetición para la adición del anticuerpo a los pozos, el conjugado enzimático, el sustrato y la solución de paro. Pipeta multicanal. Pipeta de ocho canales con 250µL de capacidad por canal y puntas de pipeta de polipropileno. Se recomienda para la adición a los pozos de la solución de lavado. Pipeta para carga de pozos. Pipetas manuales ajustables o de volumen fijo con capacidad de 50µL y puntas de pipeta recomendadas por el fabricante. Se recomienda para la adición a los pozos de los estándares, las muestras de campo y las muestras de control de calidad. Recipiente para la solución de lavado. Recipiente de plástico diseñado para pipetas multicanal (VWR no. 21007-970). Se recomienda que tenga una capacidad de 55 mL. Viales de cuatro mililitros. Pueden utilizarse viales de vidrio de borosilicato, con tapas que contengan empaques de PTFE. Se recomiendan para colocar y almacenar el filtrado de la muestra después de la lisis. Viales de quince a cuarenta mililitros. Los viales utilizados deben de ser de vidrio de borosilicato, transparentes o ámbar, con tapas con empaques de PTFE. Se recomiendan viales con capacidad en este rango para el procedimiento de lisis. No deben utilizarse botellas tratadas en un horno de mufla a alta temperatura como procedimiento de limpieza. B.1.1.6 Reactivos y soluciones Agente reductor de cloro residual. El tiosulfato de sodio (no. CAS 7772-98-7) se utiliza para reducir el cloro residual en las muestras de agua potable en el momento de la recolección. Estándares de calibración. Se recomienda el uso de los estándares de calibración suministrados en los kits ELISA. Se permiten los estándares de calibración preparados en el laboratorio. Los laboratorios deben utilizar prácticas apropiadas de control de calidad para determinar cuándo deben reemplazarse los estándares. Solvente regular y para PDS. El metanol (no. CAS 67-56-1) o el indicado por el fabricante; se usa para reconstituir MC-LR si este material se adquiere como un sólido, y para diluir el material de MC-LR para preparar soluciones PDS. Soluciones y PDS de MC-LR. Se recomienda obtener la MC-LR (no. CAS 101043-37-2) como una solución con una concentración de al menos 10 µg/mL o como el material puro. En caso de que sea como material puro, este debe de reconstituirse en metanol para obtener una solución con una concentración de al menos 10 µg/mL. Esta solución de MC-LR debe diluirse con metanol para preparar soluciones PDS. Las concentraciones de MC-LR para soluciones PDS deben de ser seleccionadas de tal manera que al menos se utilicen 5µL para fortificar muestras de control de calidad o para preparar estándares de calibración. Más de una concentración de solución PDS puede ser necesaria para cumplir con este requisito. B.1.1.7 Procedimiento B.1.1.7.1 Recolección, conservación y almacenamiento de muestras Antes del salir a colectar las muestras, debe agregarse tiosulfato de sodio a cada botella de muestra. La concentración final de tiosulfato sódico en la muestra debe ser de 100 mg/L. El tiosulfato de sodio no debe de diluirse en agua al preparar las botellas para las muestras. El agente reductor debe añadirse a la botella vacía en forma sólida. En el campo, abra el grifo y deje que el sistema se descargue durante aproximadamente 5 minutos. Llenar cada botella, teniendo cuidado de no eliminar el tiosulfato de sodio, e invertir varias veces para mezclar la muestra con el agente reductor. Colecte suficiente muestra para cumplir con los requisitos de este método. Para el tamaño de la muestra debe de considerarse un volumen suficiente para preparar las muestras de control de calidad y el volumen apropiado para el almacenamiento congelado. No debe utilizarse ácido ascórbico para reducir el cloro en las muestras de agua de uso y consumo humano. Durante los estudios para evaluar la estabilidad de las microcistinas durante el transporte y el almacenamiento, se ha descrito que éstas se degradan en presencia de ácido ascórbico. La adición de tiosulfato de sodio no es necesaria para agua de uso y consumo humano antes del tratamiento (muestras colectadas antes de la entrada al sistema de tratamiento), sin embargo, puede añadirse si el laboratorio elige preparar un solo tipo de contenedor de muestra. Las muestras deben ser refrigeradas durante el envío y no deben exceder los 10 °C durante las primeras 48 horas después de la recolección. Se debe confirmar que las muestras están por debajo o igual a 10 °C cuando se reciben en el laboratorio. Una temperatura superior a 10 °C es aceptable en aquellos casos en los que el tiempo de traslado es corto y las muestras no tienen tiempo suficiente para alcanzar la temperatura mencionada. En este caso, deben revisarse las bolsas de hielo en las hieleras de transporte. Si estas aún permanecen congeladas, las muestras pueden considerarse válidas. Las muestras deben de ser congeladas al llegar al laboratorio. Para muestras de agua de uso y consumo humano que ha pasado a través de un sistema de tratamiento, debe analizarse una muestra de cada hielera de transporte utilizando ensayos comunes para determinar el cloro libre residual, por ejemplo, la técnica colorimétrica N, N-dietil-p-fenilendiamina (DPD). La concentración total de cloro debe ser menor que el límite de detección del ensayo. Se puede recoger una muestra duplicada para llevar a cabo el ensayo de cloro libre residual. Para el almacenamiento en congelación, deben utilizarse botellas de vidrio de borosilicato, claras o ámbar, con tapas con empaques de PTFE. Seleccione la capacidad de la botella y el volumen de la muestra para evitar la ruptura de las botellas durante la congelación. Planee con anticipación para contar con suficiente volumen para preparar muestras de control de calidad como se requiere en este método. Las muestras deben de ser analizadas lo antes posible. Las muestras que se colectan y almacenan deben analizarse dentro de los 14 días posteriores a la recolección. Este periodo de 14 días se estableció como un tiempo de retención seguro basado en la evidencia empírica: durante el desarrollo del método, se observó la degradación de microcistinas en dos matrices de agua potable después de tres semanas de almacenamiento en congelación. Las muestras pueden filtrarse y analizarse en cualquier momento después de lisis, dentro de los 14 días posteriores a la recolección. Si no se analizan inmediatamente, las muestras lisadas pueden almacenarse mediante congelación en viales de vidrio con tapas con empaques de PTFE, por ejemplo, 1 ml de muestra lisada y filtrada contenida en un vial de 4 ml. B.1.1.7.2 Procedimiento analítico Este apartado describe el procedimiento para preparar las muestras y procesar la placa de microtitulación para realizar el ELISA. Fortifique las muestras QC antes de la etapa de lisis (LFB, LFSM y LFSMD). Si las muestras se congelaron, es aceptable fortificar después de este primer ciclo de lisis. Mezclar completamente e inmediatamente transferir 5 a 10 mL de cada muestra de campo en un vial de 40 mL para comenzar tres ciclos de congelación-descongelación. Si la muestra se congeló previamente, sólo se necesitan dos ciclos de congelación-descongelación. Pueden utilizarse viales más pequeños, pero reduzca el volumen de la muestra a menos del 25% de la capacidad del vial. Asegúrese de que las muestras estén completamente congeladas y completamente descongeladas durante cada ciclo. Descongelar las muestras a aproximadamente 35 °C en un baño de agua y mezclar después de cada ciclo. Filtrar 1 a 2 mL de cada muestra lisada en un vial de 4 mL usando un filtro de jeringa de fibra de vidrio. Para realizar el ELISA siga las instrucciones del fabricante para agregar muestras y reactivos a la placa. Llene dos pozos con cada estándar de calibración, muestra de campo y muestra de control de calidad. Al construir el lote de análisis debe asegurarse de que se incluyen todos los elementos de control de calidad requeridos. Las muestras de agua de uso y consumo humano pueden ser procesadas en el mismo lote de análisis. Para el desarrollo del color, si se procesan las placas manualmente, utilice una técnica que asegure que cada pozo se incube con sustrato exactamente durante el mismo período. Para lograr esto, añadir solución de paro en la misma secuencia y al mismo ritmo que la adición de sustrato. Medir el color leyendo la absorbancia a 450 nanómetros usando un lector de placas de microtitulación. B.1.1.7.3 Análisis de datos y cálculos Se debe utilizar un ajuste de curva logística de cuatro parámetros. No se permiten otros modelos de ajuste de curvas. Calcular la concentración de la muestra para cada pozo utilizando la calibración multipunto. Para cada campo y muestra QC, debe promediarse los dos valores de concentración de cada pozo. Utilice esta promedio para reportar los resultados de las muestras y para evaluar los resultados del control de calidad frente a los límites de aceptación. Reporte solamente los valores que están entre el MRL y el estándar de calibración más alto. Los resultados finales se redondearán a dos cifras significativas. Si un resultado excede el rango de la curva de calibración, diluya la muestra con agua grado reactivo. Basándose en la concentración estimada, seleccione un factor de dilución que resulte en una concentración de muestra diluida cerca de la EC50 de la curva de calibración. La concentración en la muestra diluida debe situarse entre el MRL y el estándar de calibración más alto. Analizar la muestra diluida en un lote de análisis posterior. Incorporar el factor de dilución en los cálculos de la concentración final. Informe el factor de dilución con el resultado de la muestra. B.1.1.7.4 Informe de prueba Reportar microcistinas y nodularinas totales como Ausente o Presente. En caso de que el resultado sea ausencia de microcistinas o nodularinas totales no será necesario llevar a cabo ningún método confirmatorio adicional. En caso de que el resultado sea presencia de microcistinas o nodularinas totales deberá de llevarse a cabo alguno de los dos métodos cuantitativos confirmatorios contenidos en los puntos B.1.2 y B.1.3 de este Apéndice. B.1.1.8 Control de calidad, calibración e interferencias B.1.1.8.1 Control de calidad (QC) Los requisitos de control de calidad incluyen los elementos IDC y QC asociados con cada lote de análisis. Este apartado describe cada parámetro QC, su frecuencia requerida y los criterios de desempeño que deben cumplirse para satisfacer los objetivos de calidad de datos. Estos requisitos de QC se consideran como lo mínimo aceptable en un protocolo de calidad. Sin embargo, los laboratorios pueden establecer prácticas adicionales de control de calidad para satisfacer sus necesidades específicas. El IDC debe llevarse a cabo antes de analizar las muestras de campo. El IDC incluye cuatro determinaciones: demostración de exactitud y precisión, demostración de la presencia de interferencias en el sistema, confirmación de MRL y una verificación de segunda fuente de los estándares de calibración (muestra de control de calidad). Los requisitos de IDC se describen en la Tabla B.1.1-1 de este Apéndice. Tabla B.1.1-1. Requisitos de control de calidad de la demostración inicial de capacidad (IDC)
Al realizar el IDC, el analista debe cumplir con los requisitos de calibración especificados en el apartado correspondiente. El estándar de calibración más bajo utilizado para establecer la calibración inicial debe ser igual o inferior a la concentración que representa el MRL. Las cuatro determinaciones necesarias para completar el IDC pueden incluirse en un lote de análisis único, es decir, procesarse en una única placa de ELISA. Para la demostración de exactitud y precisión, deben prepararse siete LFB repetidos fortificando cada uno con 0.50µg/L de MC-LR. Se debe añadir tiosulfato de sodio. Lisar, filtrar y analizar los LFB en un solo lote de análisis. Para este lote de análisis, se deben de incluir los LRB para cumplir con el requisito QC de LRB. También se requiere un LOW-CV. El porcentaje de desviación estándar relativa (%RSD) para MC-LR en las siete repeticiones de LFB debe ser menor o igual a 15%. La recuperación media de las siete repeticiones debe ser mayor o igual que 70% e inferior o igual a 130%.   Para la demostración de la presencia de interferencias en el sistema, en el mismo lote de análisis llevado a cabo para la exactitud y precisión en la IDC, lisar, filtrar y analizar cinco LRB. Los LRB deben contener tiosulfato de sodio. Los LRB deben distribuirse a lo largo de toda la placa. El resultado obtenido para cada LRB debe ser inferior a la media del MRL. Para la confirmación del MRL, se debe establecer una concentración objetivo para el MRL en función del uso destinado del método. Confirme el MRL siguiendo el procedimiento descrito a continuación: Preparar y analizar muestras de MRL - Fortifique siete LFB replicados con MC-LR en, o por debajo, la concentración de MRL propuesta. Los LFB deben contener tiosulfato de sodio como se especifica. Lisar, filtrar, y analizar las muestras en un lote de análisis único. El lote de análisis debe incluir dos LRB y un LOW-CV. Calcular las estadísticas del MRL - Calcular la media y la desviación estándar de las siete réplicas. Determinar el intervalo medio para el intervalo de predicción de resultados (HRPIR) usando la siguiente ecuación: 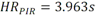 En donde: s desviación estándar 3.963 valor constante para 7 réplicas Calcular los límites superiores e inferiores para el intervalo de predicción de resultados (PIR=promedio±HRPIR) como se muestra a continuación:   Estas ecuaciones se definen sólo para siete muestras repetidas. Para los criterios de aceptación de MRL, se confirma el MRL si el límite superior del PIR es menor o igual a 150% y el límite inferior del PIR es mayor o igual al 50%. Si estos criterios no se cumplen, el MRL puede establecerse demasiado bajo y la confirmación debe repetirse, o se debe establecer y confirmar el MRL a una concentración más alta. Para las QCS, debe analizarse una QCS de nivel medio, para confirmar la exactitud de los estándares de calibración primaria. Los elementos QC que deben incluirse en cada lote de análisis. Los requisitos de lotes de análisis de QC se resumen en la Tabla B.1.1-2, de este Apéndice. Tabla B.1.1-2. Requisitos del lote de análisis de QC
Todas las muestras de campo y de QC deben añadirse a por lo menos dos pozos (pozos réplica). El %CV de los valores de absorbancia determinados para los pozos réplica debe ser menor o igual a 15%. Calcular el % CV como sigue: 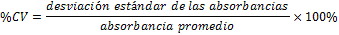 Si el %CV excede 15% de la muestra de campo o la muestra de QC (LOW-CV, LRB, LFB, LFSM y LFSMD), entonces la muestra no se considera válida. Note que los pozos réplica de los estándares de calibración deben cumplir un conjunto de criterios diferentes para %CV. Para cada lote de análisis, preparar, lisar y filtrar un Blanco de reactivo de laboratorio (LRB). El LRB debe contener tiosulfato de sodio si muestras de agua potable se incluyen en el lote de análisis. Analizar el LRB por duplicado colocando un par de pozos réplica en lados opuestos de la placa (cuatro pozos totales). La concentración total de microcistina y nodularina en cada LRB debe ser inferior a la mitad del MRL. Si la concentración es igual o mayor que este nivel, entonces cualquier muestra con un resultado positivo en el lote de análisis no se considera válida. No se permite restar valores del blanco de los resultados de la muestra. Con cada lote de análisis, debe analizarse un estándar de verificación de calibración de bajo alcance (LOW-CV). El LOW-CV es un estándar de calibración preparado a una concentración igual o inferior al MRL. Puede utilizarse un estándar de calibración del kit. No debe añadirse tiosulfato de sodio, no lisar y no filtrar el LOW-CV. La concentración analizada en el LOW-CV debe ser mayor o igual al 50% y menor o igual al 150% del valor real. Si el resultado no cumple este criterio, entonces todo el lote de análisis no es válido. Se requieren al menos dos blancos fortificados de laboratorio (LFB), a una concentración idéntica, con cada lote de análisis. Añadir tiosulfato de sodio si muestras de agua potable se incluyen en el lote de análisis. Fortificar el LFB cerca de la EC50 de la curva de calibración. Lisar y filtrar cada LFB en un vial por separado. Como criterio de aceptación para el LFB, el porcentaje de recuperación para cada LFB debe ser mayor o igual a 60% y menor o igual a 140% del valor real. Si el LFB no cumple este criterio, entonces todo el lote de análisis no se considera válido. Se requiere un conjunto de matriz de muestra fortificada en laboratorio (LFSM) y duplicado de la matriz de muestra fortificada en laboratorio (LFSMD) con cada lote de análisis en una muestra de agua de uso y consumo humano, sin embargo, si más de 20 muestras de agua están presentes en el lote de análisis, entonces se requieren dos conjuntos. De la misma manera, se requiere un conjunto LFSM y LFSMD con cada lote de análisis en una muestra de agua antes de la entrada al sistema de tratamiento, sin embargo, si hay más de 20 muestras de agua en el lote de análisis, entonces se requieren dos conjuntos. La concentración nativa en la muestra debe determinarse en una muestra de campo de manera independiente. En la rutina de trabajo, distribuya los LFSM entre las diversas fuentes de agua de uso y consumo humano (antes y después del tratamiento). Se requieren tres alícuotas separadas de una muestra de campo para determinar la concentración nativa y para preparar el LFSM y el LFSMD. Preparar el LFSM y LFSMD fortificando dos alícuotas de la misma muestra de campo con una cantidad apropiada de MC-LR. Si está congelado, descongelar las muestras. Mezclar bien para homogeneizar la muestra antes de distribuir en los tres viales. Elija una concentración tal que el resultado de la microcistina total y la nodularina se encuentre cerca de la EC50 de la curva de calibración y fortifique al menos el doble de la concentración nativa, si se conoce. Lisar y filtrar las muestras, o si las muestras fueron congeladas, completar dos ciclos más de lisis. Si no se conoce la concentración en este tipo de muestras de agua, seleccione aleatoriamente las muestras y fortifique con aproximadamente 1.0µg/L de MC-LR. Si la concentración inicial en los LFSM seleccionados al azar es alta, el resultado de la muestra fortificada puede caer fuera del intervalo de la calibración de ELISA, o fallar en cumplir el requisito de fortificar a una concentración al menos dos veces del valor nativo. En estos casos, los resultados del control de calidad se consideran inutilizables y pueden descartarse. Sin embargo, el laboratorio debe intentar recolectar datos válidos de LFSM con el tiempo para este tipo de muestras de agua. Cálculo del porcentaje promedio de recuperación - Calcular el porcentaje promedio de recuperación (%R) para cada conjunto de LFSM y LFSMD utilizando la siguiente ecuación: 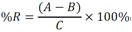 En donde: A concentración determinada promedio del LFSM y LFSMD B concentración determinada en la muestra sin fortificar C concentración fortificada Para obtener resultados significativos del porcentaje de recuperación, se debe corregir el valor promedio del LFSM y LFSMD para la concentración nativa en la muestra no fortificada, incluso si el valor nativo es menor que el MRL. El porcentaje promedio de recuperación para cada conjunto LFSM y LFSMD debe ser mayor o igual al 60% y menor o igual al 140% del valor real. Si el porcentaje de recuperación queda fuera de este intervalo, y el rendimiento del laboratorio está en control de los LFB dentro del mismo lote de análisis, la recuperación puede tener un sesgo de matriz. Calificar el resultado para la muestra de la cual se preparó el LFSM como "matriz sospechosa". Calcular la diferencia porcentual relativa (RPD) usando la siguiente ecuación: 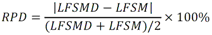 El RPD para cada conjunto de LFSM y LFSMD debe ser menor o igual al 40%. Si la RPD cae fuera de este intervalo y el rendimiento del laboratorio está en control de los LFB dentro del mismo Lote de Análisis, la precisión puede tener un sesgo de matriz. Calificar el resultado para la muestra de la cual se preparó el LFSMD como "matriz sospechosa". Una QCS debe ser analizada durante el IDC y posteriormente con cada nuevo lote de estándares de calibración. La MC-LR utilizado para el QCS debe obtenerse de una fuente independiente de la fuente de los estándares de calibración. Preparar el QCS en agua grado reactivo cerca de la EC50. Se puede utilizar un QCS suministrado con el kit ELISA si este criterio es conocido. El porcentaje de recuperación de MC-LR en el QCS debe ser mayor o igual que 70% e inferior o igual a 130%. B.1.1.8.2 Calibración Se requiere una calibración con cada lote de análisis. Utilice las concentraciones indicadas en las instrucciones del kit. No agregue niveles de calibración adicionales ni elimine ningún nivel. Los laboratorios pueden preparar estándares de calibración, sin embargo, el número de niveles y concentraciones debe coincidir con los del kit original. Cada estándar de calibración debe agregarse en al menos dos pozos. El estándar de calibración más bajo debe ser igual o inferior a la concentración del MRL. La curva de calibración se valida evaluando el %CV de los valores de absorbancia para los pozos réplica que representan cada nivel de calibración y el coeficiente de correlación de la curva logística de cuatro parámetros. Calcular el %CV para cada uno de los valores de absorbancia pareados, incluido el estándar "cero". El %CV para cada par debe ser menor o igual al 10%. Sin embargo, se permite que un par exceda del 10% siempre que el %CV sea menor o igual al 15%. El cuadrado del coeficiente de correlación (r2) de la curva de cuatro parámetros debe ser mayor o igual a 0.98. Si la calibración se encuentra fuera de los límites %CV o el valor r2 es inferior a 0.98, entonces todo el lote de análisis no se considera válido. Analizar las muestras en un lote de análisis posterior. Congelar las muestras filtradas si este lote de análisis no se puede completar el mismo día que el ensayo original. Cada muestra debe estar dentro del tiempo de espera de 14 días para repetir el ensayo. B.1.1.8.3 Interferencias La exactitud del procedimiento de ELISA depende de la técnica del analista, la precisión de los volúmenes pipeteados y períodos de incubación uniformes a través de los pozos de cada placa. La desviación del ensayo se refiere a la imprecisión sistemática en lugar de la imprecisión al azar en las concentraciones determinadas del analito de interés, cuya magnitud depende de la posición de la muestra dentro de la placa. Una posible causa de la desviación del ensayo son ligeras diferencias en los tiempos de incubación a medida que se añaden los reactivos secuencialmente a través de la placa (Davies, Chris. 2005."Concepts" en "The Immunoassay Handbook", 3rd ed.; Ed. Wild, David; Elsevier, Ltd. Oxford, UK, p 119 (sección titulada "Assay Drift")). Para detectar la desviación del ensayo es necesario distribuir a través de la placa muestras de control idénticas. Este método incluye medidas de control de calidad para evaluar la desviación del ensayo. Durante la IDC, los laboratorios deben analizar cinco LRB distribuidos a través de la placa y cada lote de análisis debe incluir dos LRB colocados en lados opuestos de la placa. Debido a que los LRB proporcionan valores de absorbancia cerca de la meseta superior de la curva de calibración, las concentraciones LRB calculadas son sensibles a ligeros cambios en la absorbancia determinada. Si los LRB distribuidos pasan el límite de control de calidad en una media del valor de MRL, entonces la desviación del ensayo de la placa es mínima y puede considerarse bajo control. Considerando que la microcistina puede ser determinada a la entrada del sistema de tratamiento, en donde el agua puede provenir de una fuente natural, las muestras de agua colectadas en la entrada del sistema de tratamiento, para este método, podrían considerarse como muestras ambientales. En ese sentido es importante observar que durante el desarrollo del método, se ha observado un sesgo positivo de aproximadamente el 30% en muestras de agua ambientales de una sola fuente. Dos aguas ambientales de otras fuentes no causaron sesgo de matriz. Durante el desarrollo del método, se evaluaron seis fuentes de agua potable para los efectos de la matriz. De estos, tres mostraron sesgo positivo en un rango de 12 a 15%. Tres fuentes no causaron sesgo de matriz. Este método contempla el análisis de muestras colectadas tanto de agua para uso y consumo humano que ha pasado a través de sistema de tratamiento como de muestras de agua colectadas en la entrada del sistema de tratamiento que provienen de fuentes naturales (muestras ambientales). Con la finalidad de evitar la contaminación cruzada, es importante separar las jeringas de vidrio utilizadas para filtrar el agua de muestras ambientales, que puede contener altos niveles de microcistinas, de las usadas para filtrar el agua potable de uso y consumo humano que ha pasado a través de sistemas de tratamiento. Alternativamente, pueden utilizarse jeringas de plástico desechables. Es importante limpiar adecuadamente los recipientes de las muestras que son vidrio en caso de ser reutilizados. No deben de reutilizarse septos de botellas que contengan muestras de agua ambientales. Los resultados reportados por este método representan el total de las microcistinas y nodularinas basadas en el método de ELISA Adda calibrado con MC-LR. B.1.1.9 Seguridad, prevención de contaminación y manejo de residuos Cada reactivo utilizado en estos procedimientos debe ser tratado como un riesgo potencial para la salud y la exposición a estos materiales debe ser minimizada. Cada laboratorio es responsable de mantener un conocimiento de las regulaciones respecto a la manipulación segura de cualquier producto químico usado en este método. Debe ponerse a disposición de todo el personal involucrado en el análisis las hojas de datos de seguridad de los productos químicos. El riesgo principal cuando se realizan los procedimientos en este método es la exposición a cianotoxinas en las muestras y en los estándares concentrados. Debe de utilizarse equipo de protección personal apropiado para el manejo de muestras y estándares. Para obtener información sobre la prevención de contaminación aplicable a las operaciones de laboratorio descritas en este método, puede consultar referencias especializadas (p.ej. American Chemical Society, 2002). Es necesario que el laboratorio cumpla con las regulaciones vigentes y aplicables en materia de manejo de residuos ante la autoridad correspondiente. B.1.1.10 Referencias · American Chemical Society. 2002. "Less is Better, Guide to Minimizing Waste in Laboratories". · Davies, Chris. 2005. "Concepts" en "The Immunoassay Handbook", 3rd ed.; Ed. Wild, David; Elsevier, Ltd. Oxford, UK, p 119 (sección titulada "Assay Drift"). · Fischer, Werner J. et al. 2001 Congener-Independent Immunoassay for Microcystins and Nodularins. Environ. Sci. Technol.35: 48494856 · Maciel, Robert J. 1985. Standard Curve Fitting in Immunodiagnostics: a Primer. Journal of Clinical Immunoassay. Vol. 8, 98106. · Ohio EPA Division of Environmental Services. 2015. Ohio EPA Total (Extracellular and Intracellular) Microcystins ADDA by ELISA Analytical Methodology; Method 701.0 Version 2.2 (and previous versions); Ohio EPA: Reynoldsburg, OH. · Sasaki, Diane and Mitchell, Robert A. 2016. How to Obtain Reproducible Quantitative ELISA Results. Oxford Biomedical Research website. · U.S. EPA. 2004. Statistical Protocol for the Determination of the Single-Laboratory Lowest Concentration Minimum Reporting Level (LCMRL) and Validation of Laboratory Performance at or Below the Minimum Reporting Level (MRL); EPA 815-R-05-006; Office of Water: Cincinnati, OH. · U.S. EPA. 2010. Technical Basis for the Lowest Concentration Minimum Reporting Level (LCMRL) Calculator; EPA 815-R-11-001; Office of Water: Cincinnati, OH. · U.S. EPA. 2016. Determination of total Microcystints and Nodularins in Drinking Water and Ambient Eater by Adda Enzyme-Linkes Immunosorbent Assay. Method 546. B.1.2 MÉTODO PARA LA DETERMINACIÓN DE MICROCISTINAS Y NODULARINAS EN AGUA DE USO Y CONSUMO HUMANO POR EXTRACCIÓN EN FASE SÓLIDA Y CROMATOGRAFÍA LIQUIDA/ ESPECTOMETRÍA DE MASAS EN TÁNDEM (LC/MS/MS) B.1.2.1 Definiciones y términos Analito surrogado (SUR), al producto químico puro que se asemeja químicamente a los analitos de interés y es extremadamente improbable que se encuentre en cualquier muestra. Este producto químico se añade a una alícuota de muestra en una cantidad o cantidades conocidas antes del procesamiento y se mide con los mismos procedimientos usados para medir los otros analitos. El propósito del SUR es monitorear el desempeño del método con cada muestra. Blanco de reactivo de laboratorio (LRB), a la alícuota de agua grado reactivo u otra matriz blanco que se trata exactamente como una muestra incluyendo la exposición a toda la cristalería, equipo, disolventes y reactivos, conservantes de muestras y surrogados que se utilizan en el lote de análisis. El LRB se utiliza para determinar si los analitos de interés u otras interferencias están presentes en el entorno del laboratorio, los reactivos o los aparatos. Blanco fortificado de laboratorio (LFB), al volumen de agua grado reactivo u otra matriz blanco de la que se conocen cantidades del analito y todos los compuestos se añaden en el laboratorio. El LFB se analiza exactamente como una muestra y su propósito es determinar tanto si la metodología está en control como si el laboratorio es capaz de realizar mediciones precisas y exactas. Concentración más baja del nivel mínimo reportado (LCMRL), a la concentración más baja verdadera para la cual se espera una recuperación futura espera entre 50 y 150% de recuperación, con un 99% de confianza. Disociación activada por colisiones (CAD), al proceso de convertir la energía de traslación del ion precursor en energía interna por colisiones con moléculas de gas neutro para producir la disociación en los iones del producto. Duplicados de campo (FD1, FD2), a las dos muestras separadas recolectadas al mismo tiempo y el mismo lugar bajo circunstancias idénticas, y tratadas exactamente igual bajo los mismos procedimientos de campo y laboratorio. El análisis de FD1 y FD2 dan una medida de la precisión asociada con la recolección, conservación y almacenamiento de las muestras, así como de los procedimientos de laboratorio. duplicado de la matriz de muestra fortificada en laboratorio (LFSMD), al duplicado de la muestra de campo utilizada para preparar la LFSM. El LFSMD es fortificado, extraído y analizados de forma idéntica a la LFSM. El LFSMD se utiliza en lugar del duplicado de campo para evaluar la precisión del método cuando la ocurrencia de los analitos es infrecuente. Estándar de calibración (CAL), a la solución preparada a partir de la dilución primaria de la solución estándar y/o solución estándar y el surrogado. Las soluciones CAL se utilizan para calibrar la respuesta del instrumento con respecto a la concentración del analito de interés. Hoja de datos de seguridad del material (MSDS), a la información escrita proporcionada por los vendedores acerca de la toxicidad del producto químico, los riesgos para la salud, las propiedades físicas, riesgo de incendio y reactividad, incluyendo las precauciones de almacenamiento, derrame y manipulación. Ion precursor, a la molécula protonada del analito ([M + H]+ o [M + 2H]2+). En MS/MS, el ion precursor es masa seleccionada y fragmentada por CAD para producir iones producto distintivos de menor relación m/z. Ion producto, fragmentos de iones producidos en MS/MS por CAD a partir del ion precursor. Límite de detección (DL), a la concentración mínima de un analito de interés que puede ser Identificado, medido y reportado con un 99% de confianza de que la concentración del analito es mayor a cero. Esta es una determinación estadística de precisión y no se espera una cuantificación exacta a este nivel. Lote de análisis, al conjunto de muestras que se analiza en el mismo instrumento durante un período de 24 horas, incluyendo no más de 20 muestras de campo, que comienza y termina con el análisis de los estándares de verificación de calibración continua (CCC) apropiados. Pueden requerirse CCC adicionales dependiendo de la longitud del lote de análisis y/o el número de muestras de campo. Lote de extracción, al conjunto de hasta 20 muestras de campo (sin incluir las muestras de Control de Calidad, QC) extraídas juntas por la misma persona durante un día de trabajo usando el mismo lote de dispositivos SPE, disolventes, sustitutivos y soluciones para fortificar. Las muestras de QC requeridas incluyen el blanco de reactivo de laboratorio, el blanco fortificado de laboratorio, la matriz de la muestra fortificada de laboratorio y un duplicado de campo o duplicado de matriz de la muestra fortificada de laboratorio. Matriz de muestra fortificada en laboratorio (LFSM), a la muestra de campo preservada a la que se añaden en el laboratorio cantidades conocidas de los analitos de interés. La LFSM es procesado y analizado exactamente como una muestra y su propósito es determinar si la matriz de la muestra contribuye a los resultados analíticos. Las concentraciones de fondo de los analitos en la matriz de muestra deben ser determinadas en una extracción independiente de la muestra y los valores medidos en la LFSM corregidos para las concentraciones de fondo. Muestra de control de calidad (QCS), a la solución de analitos de concentraciones conocidas que se obtiene de una fuente externa al laboratorio y diferente del estándar de calibración. La segunda fuente para la solución estándar se utiliza para fortalecer el QCS a una concentración conocida. El QCS se utiliza para comprobar la integridad del estándar de calibración. Nivel mínimo reportado (MRL), a la concentración mínima que puede ser reportada como un valor cuantificado para un analito en una muestra después del análisis. Esta concentración definida no puede ser inferior a la concentración del estándar de calibración más bajo para ese analito y sólo puede utilizarse si se cumplen los criterios de control de calidad (QC) aceptables para ese estándar. Solución estándar (SSS), a la solución concentrada que contiene uno o más analitos preparada en el laboratorio utilizando materiales de referencia ensayados o adquiridos de una fuente comercial de buena reputación. Solución estándar de dilución primaria (PDS), a la solución que contiene los analitos preparados en el laboratorio a partir de soluciones estándar y diluida según sea necesario para preparar soluciones de calibración y otras soluciones de analito necesarias. Verificación de calibración continua (CCC), al estándar de calibración que contiene los analitos del método y surrogados. El CCC se analiza periódicamente para verificar la exactitud de la calibración existente para esos analitos. B.1.2.2 Símbolos y términos abreviados CAD disociación activada por colisiones CAL estándar de calibración CCC verificación de calibración continua C2D5-MC-LR microcistina LR etilada, d5 DL límite de detección ESI Ionización por electrospray FD1, FD2 duplicados de campo LCMRL concentración más baja del nivel mínimo reportado LFB blanco fortificado de laboratorio LRB blanco de reactivo de laboratorio LFSM Matriz de muestra fortificada en laboratorio LFSMD duplicado de la matriz de muestra fortificada en laboratorio MC-LR microcistina-LR mL/min mililitro/minuto [M + H]+ Primer estado de protonación del analito [M + 2H]2+ Segundo estado de protonación del analito MRL nivel mínimo reportado MSDS hoja de datos de seguridad del material PDS solución estándar de dilución primaria QC control de calidad QCS muestra de control de calidad RSD desviación estándar relativa SPE extracción en fase sólida SSS solución estándar SUR analito surrogado ± más y menos B.1.2.3 Principio Una muestra de agua de 500 ml (fortificada con un surrogado) se filtra y se colectan tanto el filtrado como el filtro. El filtro se coloca en una solución de metanol que contiene 20% de agua grado reactivo y se mantiene durante al menos una hora a -20 ºC para liberar las toxinas intracelulares de las célula de cianobacterias capturadas en el filtro. El líquido se extrae del filtro y es añadido al filtrado acuoso de 500mL. La muestra de 500 ml (más la solución de toxina intracelular) es pasa a través de un cartucho de SPE para extraer los analitos de interés y el surrogado. Los analitos se eluyen de la fase sólida con una pequeña cantidad de metanol que contiene un 10% de agua grado reactivo. El extracto es concentrado a sequedad por evaporación con nitrógeno en un baño de agua caliente y luego se ajusta a un volumen de 1 ml con metanol que contiene un 10% de agua grado reactivo. Se hace una inyección de 10 µL en una LC equipada con una columna C8 interconectada a un MS/MS. Los analitos se separan e identifican comparando los espectros de masas y los tiempos de retención con los espectros de referencia y los tiempos de retención de los estándares de calibración realizados bajo condiciones idénticas de LC/MS/MS. La concentración de cada analito de interés se determina mediante calibración estándar externa. B.1.2.4 Alcance y aplicación Este es un método de cromatografía liquida/espectrometría de masas en tándem (LC/MS/MS) para la determinación de microcistinas y nodularinas (combinado intracelular y extracelular) en agua de uso y consumo humano. Los datos de exactitud y precisión han sido generados en agua grado reactivo y finalizados en aguas subterráneas y superficiales terminadas para los analitos listados en la Tabla B.1.2-1, de este Apéndice. Tabla B.1.2-1. Analitos que pueden ser determinados por este método
Este método debe de ser utilizado por analistas expertos en extracciones en fase sólida, funcionamiento de los instrumentos de LC/MS/MS y la interpretación de los datos asociados. En reconocimiento de los avances tecnológicos en sistemas y técnicas analíticas, se permite al laboratorio modificar las técnicas de evaporación, separación, columna LC, composición de fase móvil, condiciones LC y condiciones MS y MS/MS. Sin embargo, no deben de realizarse cambios a la recolección y conservación de muestras, a las etapas de extracción de muestras, ni a los requisitos de control de calidad. Las modificaciones deben ser consideradas sólo para mejorar el rendimiento del método. Modificaciones que son introducidas con el interés de reducir el costo o el tiempo de procesamiento de la muestra, pero dar lugar a un método más pobre no deben utilizarse. Los analitos deben estar adecuadamente resueltos cromatográficamente para permitir que el espectrómetro de masas proporcione un número mínimo de compuestos eluyendo dentro de una ventana de tiempo de retención. La sensibilidad instrumental (señal de ruido) disminuirá si se permite que demasiados compuestos eluyan dentro de una ventana de tiempo de retención. En todos los casos en los que se proponen modificaciones de del método, el analista debe realizar los procedimientos descritos en la demostración inicial de capacidad (IDC), verificar que conoce todos los criterios de aceptación del control de calidad (QC) y que el rendimiento del método aceptable puede verificarse en una matriz de muestra real. B.1.2.5 Equipos y materiales Las referencias a marcas específicas y números de catálogo se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden representar especificaciones adecuadas para los artículos. Abrazadera metálica. Abrazadera de aluminio de 47 mm (Kimble Chase #953753-0000 o equivalente). Balanza analítica. Balanza analítica capaz de pesar 0. 000 1 g Base de filtro O-ring. Anillo de sellado de PTFE /silicona (Kimble Chase # 410171-4226 o equivalente). Base de soporte. Base de soporte para filtración de vidrio fritado de 47 mm (Kimble Chase # 953752-5047 o equivalente). Boquilla para manguera. Adaptador de tubo para aparatos de filtración (Kimble Chase #736400-1413 o equivalente). Cartuchos para dispositivo de SPE. Waters Oasis HLB, 150 mg, 6cc copolimero divinilbenceno N-vinilpirrolidona (Waters # 186003365). Contenedores de muestra. Botellas de vidrio ámbar de 500 mL con tapa con rosca de politetrafluoroetileno (PTFE). Columna analítica. Columna LC C8 (2.1x100mm) cargada con partículas de fase sólida C8 de 2.6µm (Phenomenex Kinetex # 00D-4497-AN). Cualquier columna equivalente que proporcione una resolución adecuada, forma de pico, capacidad, exactitud y precisión puede ser usada. Contenedores para colectar filtrado. Botellas de vidrio ámbar de 500 ml (Fisher #02-542-4C o equivalente) y tapa de botella GL45 (Fisher #13247GL45 o equivalente; No se muestra en la figura). Dispositivo para filtración. En la Fig. B.1.2-1 de este Apéndice, se indican número de partes:  Fig. B.1.2-1 Partes del dispositivo de filtración Embudo. Embudo de vidrio de 300 mL de 47 mm (Kimble Chase # 953751-0000 o equivalente). Espectrómetro de masas en tandem (MS/MS) con sistema de datos. El espectrómetro de masas debe ser capaz de ionizar por ionización por electrospray (ESI) cerca de la tasa de flujo LC sugerida de 0.3 mL/min. El sistema debe ser capaz de realizar MS/MS, para producir iones de producto únicos para métodos de segmento de analitos de tiempo de retención especificados. Un mínimo de 10 exploraciones a través del pico cromatográfico, es necesario para asegurar una precisión adecuada. Extractor manual. Bomba manual de vacío con modelo de gran volumen Visiprep (Supelco #57030 y # 57275 o equivalente) para cartuchos de extracción. Filtro de membrana. Membrana nucleoporo de filtro de policarbonato de 47 mm con un tamaño de poro 0.4 µm (Whatman # 111107 o equivalente). Microjeringas. Los tamaños sugeridos incluyen jeringas de 5, 10, 25, 50, 100, 250, 500 y 1 000 µL. Sistema de concentración de extracto. Los extractos se concentran por evaporación con nitrógeno utilizando un baño de agua con una temperatura no mayor a 60 °C (Meyer N-Evap, Modelo 111, Organomation Associates, Inc. o equivalente) Sistema de datos. Se requiere un sistema de datos interconectado para adquirir, almacenar, reducir y producir datos espectrales de masa. El software de la computadora debe tener la capacidad de procesar datos de LC/MS/MS almacenados, reconociendo un pico de LC dentro de cualquier ventana de tiempo de retención dada. El software debe permitir la integración de la abundancia de iones de cualquier ion específico dentro del tiempo especificado o dentro del límite del número de escaneo. El software debe ser capaz de calcular los factores de respuesta relativa, construir regresiones lineales o curvas cuadráticas de calibración, y calcular las concentraciones del analito de interés. Sistema de liberación muestras. Se recomienda el uso de un sistema de tubos de transferencia (Supelco "Visiprep", # 57275 o equivalente), para transferir la muestra directamente del contenedor de muestra al cartucho de SPE. Sistema de vacío de laboratorio o aspirador. Con una capacidad suficiente para mantener un vacío de aproximadamente 10 a 15 pulgadas de mercurio para extraer cartuchos. Sistema para cromatografía líquida (LC). Instrumento capaz de inyectar de forma reproducible alícuotas de hasta 10 µL y realizar gradientes lineales binarios a un flujo constante cerca del flujo utilizado para el desarrollo de este método (0.3mL/min). El uso de un calentador de columna es opcional. Tapa de botella con agujero. Tapa GL45 para botella con orificio para la base del soporte del filtro (Kimble # 410170-4534, o equivalente). Tubos cónicos de centrífuga. Tubos cónicos de centrifugación de vidrio de 15 ml (Corning # 8082-15) u otra cristalería adecuada para recolectar el eluyente de la fase sólida después de la extracción. Tubos de cultivo de fondo redondo. Tubos de cultivo de fondo redondo de vidrio de 15 ml (Corning # 9826-16X o equivalente) u otra cristalería adecuada para su uso en la liberación de la toxina del filtro. Viales de automuestreo. Viales de vidrio ámbar de automuestreo de 2 mL (National Scientific #C4000-2W o equivalente) con tapa y septos de PTFE (National Scientific # C4000-53 o equivalente). B.1.2.6 Reactivos y soluciones Las referencias a marcas específicas y números de catálogo se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden representar especificaciones adecuadas para los artículos. Ácido ethilendiaminotetraacetico, sal trisódica. (Trisodium EDTA, no. CAS 10378-22-0). Inhibe la hidrólisis de los analitos catalizada por metales. La sal trisódica se utilizada en lugar de la sal disódica dado que el pH de la solución salina trisódica es más próximo al pH deseado de 7 (Sigma #ED3SS o equivalente). Reactivo para preservación de la muestra. Dado que es sólido a temperatura ambiente, puede añadirse a la botella para la muestra antes de salir a colectar la muestra al campo. Ácido L-ascórbico. (no. CAS 50-81-7). Reduce el cloro libre en el momento de recolección de la muestra. (Sigma-Aldrich #255564 o equivalente). Reactivo para preservación de la muestra. Dado que es sólido a temperatura ambiente, puede añadirse a la botella para la muestra antes de salir a colectar la muestra al campo. Agua grado reactivo. Agua purificada que no contiene ninguna cantidad de sustancia medible de cualquier analitos o compuesto interferente mayor de 1/3 del MRL para cada analito de interés. Argón. Utilizado como gas de colisión durante experimentos de MS/MS. El argón debe cumplir o exceder las especificaciones del fabricante del instrumento. El gas nitrógeno puede usarse como gas de colisión siempre que se obtenga una sensibilidad suficiente (formación de iones de producto). Cristales Trizma preempaquetados. Grado de reactivo. Mezcla premezclada de Tris [Tris (hidroximetil)aminometano] y Tris HCL [Tris (hidroximetil)aminometano clorhidrato]. pH 7.0 (Sigma-Aldrich #T-7193 o equivalente) - Alternativamente, puede usarse una mezcla de los dos componentes con un relación de peso 153.5/1 Tris HCl/Tris. Ambas mezclas son usadas para producir un pH cercano a 7.0 a 25°C en agua grado reactivo. Trizma funciona como una solución amortiguadora. Reactivo para preservación de la muestra. Dado que es sólido a temperatura ambiente, puede añadirse a la botella para la muestra antes de salir a colectar la muestra al campo. Estándares de calibración (CAL). Preparar una serie de al menos cinco concentraciones de soluciones de calibración en metanol que contiene 10% de agua, a partir de las diluciones de la PDS del analito. Las concentraciones sugeridas son una descripción de las concentraciones usadas durante el desarrollo del método y pueden ser modificadas de acuerdo con la sensibilidad del equipo. Los rangos de concentración utilizados durante el desarrollo del método fueron 10-400 µg/L, excepto para MC-RR (4.7-187.5 µg/L), nodularina-R (4.9 - 195.7 µg/L) y MC-LA (25 - 1 000 µg/L). Los rangos de concentración más grandes requerirán más puntos de calibración. El SUR se agrega a los estándares CAL en una concentración constante. Durante el desarrollo del método, la concentración del SUR fue de 129.8 µg/L en el estándar (259.6 ng/L en la muestra acuosa). La concentración más baja del estándar CAL debe estar en o por debajo del MRL, lo cual depende de la sensibilidad del sistema. Los estándares CAL pueden usarse también como CdC. Durante el desarrollo del método, los estándares CAL se encontraron estables durante dos semanas si se almacena a -4°C o menos. Tiempos más largos de almacenamiento son aceptables siempre y cuando se documenten las medidas de control de calidad demostrando la estabilidad del CAL. Formiato de amonio. (CH5O2N, no. CAS 540-69-2) - Alta pureza, demostrando estar libre de analitos e interferencias; grado LC/MS (Fluka# 55674) o equivalente). Gases, reactivos y solventes. Deben ser usados los mejores productos de reactivos químicos, a menos que se indique otra cosa, se pretende que todos los reactivos contengan las especificaciones del Comité de Reactivos Analíticos de la Sociedad Química Americana. Se pueden utilizar otros grados de reactivo, determinando primero que el reactivo sea de pureza suficientemente alta para permitir su uso sin disminuir la calidad de la determinación. Metanol. (CH3OH, no. CAS 67-56-1) - Alta pureza, demostrando estar libre de analitos e interferencias (Fisher Optima grado LC/MS o equivalente). Nitrógeno. Ayuda a la generación de aerosoles y a la desolvatación del aerosol líquido en la Ionización por electrospray (ESI) y se utiliza como gas de colisión en algunos equipos MS/MS. El nitrógeno utilizado debe cumplir o exceder las especificaciones del fabricante del instrumento. Solución amortiguadora de formiato 20mM. Para preparar 1 L, añadir 1.26 g de formiato de amonio a 1 L de agua grado reactivo. Esta solución es propensa a pérdidas de volatilidad y debe ser sustituido por lo menos cada 48 horas. Soluciones estándar. Cuando la pureza evaluada de un compuesto es igual o mayor al 95%, el peso puede utilizarse sin corrección para el cálculo de concentración de las soluciones estándar. Las concentraciones sugeridas son una descripción de las concentraciones que se utilizan durante el desarrollo del método y pueden ser modificadas para ajustarse a la sensibilidad del equipo que se utilice. Las soluciones estándar para la fortificación de muestras generalmente deben ser preparadas en pequeños volúmenes, que puedan ser medidos con precisión para minimizar la adición de exceso solvente orgánico para las muestras acuosas. Los laboratorios deben utilizar prácticas de control de calidad para determinar cuándo es necesario reemplazar sus soluciones estándar. No deben utilizarse puntas de pipetas de polipropileno para la dosificación de soluciones que contiene analitos de este método, ya que se ha reportado la adsorción de microcistina por el polipropileno. Soluciones estándar del analito. Las soluciones estándar del analito pueden comprarse comercialmente como soluciones en ampolletas o preparadas a partir de material puro. Solución estándar del analito (10 - 500 µg/ml). Las cianotoxinas puras son compradas generalmente en cantidades de 10-500µg. Debido a la pequeña cantidad y a la toxicidad de estos analitos, el pesado de las cianotoxinas no es factible. Si se preparan a partir de material puro, simplemente debe añadirse 1 mL de metanol al material puro comprado (10 - 500 µg) para obtener una concentración final de 10 - 500 µg/mL. Repita para analito preparado a partir de material puro. Alternativamente, pueden comprarse soluciones estándar de los analitos preferiblemente en metanol si están disponibles en el mercado. Las soluciones estándar son estables al menos 6 meses si son almacenadas a -15°C o menos en frascos color ámbar de tapa de vidrio. Soluciones estándar del analito surrogado (SUR). El SUR para este método es la C2D5-MC-LR. Este Sur marcado isotópicamente contiene funciones similares al analito. Aunque pueden usarse soluciones estándar de SUR alternativos siempre que estén marcados isotópicamente con grupos funcionales similares al analito, el analista debe documentar las razones por las cuales utiliza estándares alternativos de SUR. Asimismo, las soluciones estándar de SUR deben de cumplir con los requisitos de control de calidad. Solución estándar de dilución primaria (PDS) (0.94 - 5.0 ng/µl). La PDS del analito contiene todos o una parte de los analitos de interés a diferentes concentraciones en metanol. La respuesta en ESI y MS/MS varía según el compuesto, por lo tanto, puede ser necesaria una mezcla de concentraciones en la PDS del analito. Durante el desarrollo del método, las soluciones PDS del analito fueron preparadas tal que aproximadamente se obtuvo la misma respuesta del equipo para todos los analitos. La PDS del analito se preparó en metanol a las concentraciones de 0.94 a 5.0 ng/µL. La PDS del analito se prepara por dilución de la combinación de las soluciones de estándar del analito de interés y se utiliza para preparar los estándares CAL y fortalecer los LFB, LFSM, LFSMD y FD con los analitos de interés. La PDS del analito ha demostrado ser estable para un mes cuando se almacena a -15 °C o menos en frascos de vidrio ámbar con tapa de rosca. Tabla B.1.2-2. Determinación de la concentración final del analito en la PDS
Solución estándar de dilución primaria del surrogado (PDS SUR; 6.49 ng/µL). La PDS del SUR es preparada por dilución de 64.9 µg de material puro en 10 mL de metano. Esta solución se utiliza para fortificar todas las soluciones de control de calidad y las muestras de campo. Se ha demostrado que el PDS es estable durante al menos un mes cuando se almacena a -15 °C o menos. Utilice 20 µL de este PDS SUR con 6.49 ng/µL para fortificar las soluciones acuosas de control de calidad de 500 mL y las muestras de campo antes de la extracción. Esto producirá una concentración de 259.6 ng/L del SUR en las soluciones acuosas de QC y en las muestras de campo. La concentración del SUR puede ajustarse para adaptarse a sensibilidad del equipo. 2-cloroacetamida. (no. CAS 79-07-2) - Inhibe el crecimiento microbiano y la degradación del analito (Sigma-Aldrich # C0267 o equivalente). Reactivo para preservación de la muestra. Dado que es sólido a temperatura ambiente, puede añadirse a la botella para la muestra antes de salir a colectar la muestra al campo. B.1.2.7 Procedimiento B.1.2.7.1 Recolección, conservación y almacenamiento de muestras Recolectar las muestras en botellas de 500 mL de vidrio color ámbar con tapones de rosca con revestimiento de teflón. No utilice botellas para muestras mayores a 500 mL (dado que los pasos de enjuague no son óptimos para tamaños grandes de botella). Tamaños de muestra más pequeños pueden utilizarse ya que el MRL puede ser resuelto, sin embargo, no deben de ser menores a 100 mL. Todo el volumen completo de la muestra en la botella debe ser utilizado (por ejemplo, una alícuota de 100 mL no debe tomarse de una botella de 500 mL porque por que la botella de la muestra debe ser enjuagada). Los siguientes reactivos para la conservación de la muestra, listados en la Tabla B.1.2-3, de este Apéndice, son añadidos a cada botella de muestra como un sólido antes de su envío al campo o antes de la colecta de la muestra. Tabla B.1.2-3. Reactivos para conservación de la muestra
Para la recolección de la muestra debe abrir el grifo de agua fría y dejar fluir el sistema hasta que la temperatura del agua se haya estabilizado (aproximadamente 3 a 5 minutos) y entonces debe de tomar la muestra. Las botellas deben llenarse, teniendo cuidado de no eliminar los reactivos de preservación de la muestra. Las muestras no necesitan ser colectadas sin que quede espacio libre en el vial o botella de la muestra. Después de recolectar la muestra, tapar la botella y agitar con la mano hasta que el reactivo para la conservación es disuelto. Tenga en cuenta que el 2-cloroacetamida es lento en disolverse especialmente en agua fría. Mantener la muestra sellada desde el momento de recolección hasta la extracción. Las muestras deben ser enfriadas durante el envío y no deben exceder los 10 °C durante las primeras 48 horas después de la recolección. Cuando las muestras son recibidas en el laboratorio debe confirmarse que la temperatura de la muestra sea igual o menor a 10 °C. Las muestras almacenadas en el laboratorio deben de conservarse a o por debajo de 6 °C hasta su extracción, pero no deben ser congeladas. Las muestras que son significativamente superiores a 10 °C, en el momento de la recolección de la muestra, deben ser puestas en hielo o refrigeradas por un período de tiempo, con el fin de enfriarlas antes del envío. Esto permitirá que se envíen con la temperatura adecuada para cubrir con los requisitos anteriores. Las muestras de agua deberían ser extraídas tan pronto como sea posible después de la recolección, sin embargo, pueden extraídas dentro de 28 días posteriores a la recolección de la muestra. Los extractos deben ser almacenados en -4 °C y analizados dentro de los 28 días siguientes a la extracción. Los tiempos de espera para muestra y extracto con el porcentaje de recuperación (% prom REC) y el porcentaje de desviación estándar relativa (%RSD) se presentan en las Tablas B.1.2-4 y B.1.2-5, de este Apéndice.
B.1.2.7.2 Procedimiento analítico Este procedimiento se puede realizar manualmente o de forma automatizada mediante un robot o dispositivo de preparación de muestras automático. Puede utilizarse un sistema automático/robótico de preparación de muestras, diseñado para usarse con cartuchos de SPE siempre y cuando cumpla con todos los requisitos de control de calidad. Si se utiliza un sistema automatizado para preparar muestras, debe seguir las instrucciones de fabricante, pero todos los pasos de extracción y la elución deben ser los mismos que en el procedimiento manual. Los pasos de extracción o elución no pueden ser cambiados u omitidos para adecuar el uso de un sistema automatizado. Si se utiliza un sistema automatizado, los LRB deben rotarse entre los puertos para asegurar que todas las válvulas y la tubería cumplan los requisitos de LRB. Los cartuchos de SPE que se mencionan, están diseñados como elementos de uso individual y deben desecharse después del uso. No pueden ser restaurados o reacondicionados para su reutilización en análisis posteriores. Las muestras son conservadas, recogidas y almacenadas tal como se mencionó anteriormente. Todas las muestras de campo y de QC, incluyendo el LRB y el LFB, debe contener los conservantes listados en la Tabla B.1.2-3, de este Apéndice. Antes de la extracción, se debe verificar que el pH de la muestra es 7±0.5. Si el pH de la muestra no cumple con este requisito, la muestra debe desecharse. Si el pH de la muestra es aceptable, se debe proceder con el análisis. Antes de la extracción, marque el nivel de la muestra en el exterior de la botella de muestra para posteriormente determinar el volumen de muestra. Si se usa el peso para determinar el volumen, debe pesarse la botella con la muestra recolectada antes de la extracción. Como se mencionó, se pueden utilizar tamaños de muestra menores para obtener el MRL. Debe usarse el mismo tamaño de muestra para el LFB, FD, LFSM y LFSMD así como para la muestra de campo y todas las muestras de QC las cuales deben cumplir el tamaño de muestra más pequeño. A cada muestra a ser extraída debe añadirse alícuota de la PDS SUR, tapar e invertir para mezclar. Durante el desarrollo del método, se añade una alícuota de 20 µL de la solución PDS SUR 6.49 ng/µL a 500 mL para una concentración final de 259.6 ng/L en la muestra acuosa. Además del SUR y los conservantes, si la muestra es LFB, FD, LFSM o LFSMD, agregar la cantidad necesaria de la PDS del analito, tapar e invertir cada muestra para mezclar. Para la liberación de la toxina intracelular debe filtrarse la muestra de agua de 500 mL usando un filtro Nuclepore con el lado brilloso hacia arriba y recoger el filtrado en una botella de vidrio color ámbar de 500 mL2.1) para la extracción. Enjuagar la botella de la muestra con 5 mL de metanol que contiene un 10% de agua grado reactivo y verter el agua de enjuague en el dispositivo de filtrado y combinar el agua de enjuague con la muestra de agua filtrada. Enjuagar los lados del embudo con otros 2.5 mL de metanol que contiene un 10% de agua grado reactivo y combinar con la muestra de agua filtrada. Utilizando pinzas de metal, retire el filtro del dispositivo de filtración y doble el filtro por la mitad (parte superior del filtro hacia adentro) mientras toca únicamente los bordes del filtro. Continúe plegando el filtro hasta que esté lo suficientemente pequeño como para caber en un tubo de vidrio. Empuje el filtro hacia la parte inferior de la probeta de cristal utilizando una pipeta de vidrio. Añadir 2 mL de metanol que contiene un 20% de agua grado reactivo al tubo de ensayo que contiene el filtro (asegurándose que el filtro se cubre con el líquido) y manualmente agitar el tubo suavemente varias veces. Coloque el tubo de ensayo que contiene los 2 mL de la solución filtrada y el filtro en un congelador a -20°C por 1 a 16 horas. No exceder de 16 horas en el congelador. Si el filtro se mantiene congelado por más de 2 horas, los 500 mL del filtrado acuoso se deben mantener refrigerados a 6°C hasta la finalización del procedimiento de liberación de la toxina. Remover el tubo de prueba del congelador, agitar suavemente un par de veces y luego extraer los 2 mL del líquido con una pipeta de vidrio. Transferir estos 2 mL de líquido a los 500 mL de agua filtrada de la muestra colectada. Enjuague el filtro y el tubo de ensayo mediante la adición de otros 2 mL de metanol que contiene un 20% de agua grado reactivo al tubo de ensayo y gírelo suavemente. Extraer los 2 mL del líquido utilizando una pipeta de vidrio y transferir estos 2 mL de líquido a los 500 mL de agua filtrada de la muestra colectada. Enjuagar el filtro una segunda vez mediante la adición de otro 1 mL de metanol que contiene un 20% de agua grado reactivo al tubo de ensayo y agitar suavemente. Extraer el 1 mL de líquido utilizando una pipeta de vidrio y transferir este 1mL del líquido a los 500 mL de agua filtrada de la muestra colectada. Agitar la muestra de 500 mL varias veces para homogeneizar la muestra. Para la limpieza y acondicionado del cartucho, no permita que el material de empaque del cartucho esté seco en cualquiera de los pasos de condicionamiento. Enjuagar cada cartucho con 15mL de metanol. A continuación, enjuagar cada cartucho con 15 mL de agua grado reactivo, sin permitir que el agua caiga por debajo del borde superior del empaque. Si el cartucho queda seco durante la fase de condicionamiento, debe comenzar de nuevo. Añadir 4 - 5 mL de agua grado reactivo a cada cartucho, colocar tubos de transferencia de muestras, encender la aspiradora y comenzar la adición de la muestra filtrada (que contienen las toxinas intracelulares liberadas) en el cartucho. Con el fin de realizar una correcta extracción de la muestra, ajuste el vacío para que el caudal aproximado sea de 10 - 15 mL/min. No permita que el cartucho esté seco antes de que toda la muestra pase a través del cartucho. Para el enjuague de la botella de la muestra y del cartucho, una vez que toda la muestra ha pasado a través del cartucho, enjuague las botellas de muestra con 10 mL de agua grado reactivo y extraiga el enjuague a través de los tubos de transferencia de la muestra y los cartuchos. Retirar los tubos de transferencia de la muestra y lavar los cartuchos con otros 5 mL de agua grado reactivo. Extraer aire o nitrógeno a través del cartucho por 10 min al alto vacío (10 - 15 pulgadas de mercurio). Para permitir la elución de la botella de la muestra y del cartucho, apague y libere el vacío. Levante la tapa del colector de extracción e inserte un bastidor con tubos de colecta en el tanque de la extracción para recolectar los extractos que se eluyen de los cartuchos. Vuelva a encender la aspiradora, pero asegúrese que el vacío no exceda de 10 de pulgadas de mercurio durante la elución. Enjuague las botellas de muestra con 5 mL de metanol que contiene 10% reactivo agua y eluir los analitos de los cartuchos tirando los 5 mL de metanol (usada para enjuagar las botellas) a través de los tubos de transferencia de la muestra y los cartuchos. Use un aspirado de baja potencia tal que el solvente salga del cartucho de gota en gota. Repita el enjuague de la botella de la muestra y la elución del cartucho con una segunda alícuota de 5 mL de metanol que contiene 10% de agua reactivo. Los volúmenes de solvente utilizados fueron optimizados para botellas de 500 mL de la muestra. El uso de botellas para muestras más grandes para las muestras de control de calidad no se recomienda dado que puede afectar negativamente la recuperación de analito. Concentrar el extracto hasta que esté seco bajo una corriente suave de nitrógeno en un baño de agua caliente (60°C). Añadir 1 ml de metanol que contiene 10% de agua reactivo al vial y agitar en vortex. Transferir una alícuota a un vial de automuestreo. Para determinar el volumen de la muestra, si el nivel de la muestra fue marcado en la botella de la muestra, utilice una probeta graduada para medir el volumen de agua necesaria para llenar la botella de la muestra original hasta la marca que hizo antes de la extracción. Determinar a los 10ml más cercanos. Si se usa el peso para determinar el volumen, pesar la botella vacía a los 10 g de peso y determinar el peso de la muestra por la substracción del peso de la botella vacía del peso de la muestra original. Asumir una densidad de 1 g/mL de muestra. En cualquier caso, el volumen de la muestra se utilizará en el cálculo final de concentración de analito. Para realizar el análisis del extracto, se deben establecer las condiciones necesarias resumidas en las Tablas B.1.2-6, B.1.2-7, B.1.2-8 y B.1.2-9, de este Apéndice. Columnas y condiciones del instrumento deben optimizarse antes de la iniciación de la IDC. Tabla B.1.2-6. Condiciones del método LC
Tabla B.1.2-7. Condiciones del método ESI-MS/MS
Tabla B.1.2-8. Origen del analito y tiempo de retención (RT) a
Tabla B.1.2-9. Condiciones del método MS/MS a
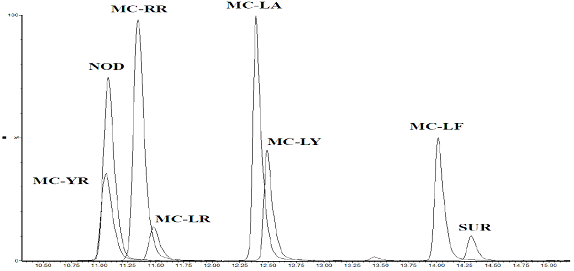 Fig. B.1.2-2 Ejemplo de cromatograma (segmentos MS/MS sobrelapados) de un estándar de calibración con el método 544 de EPA y con analitos en una concentración de 187.5-1 000 ng/L Se recomienda desviar los primeros 6 a 8 minutos del flujo LC para residuos. Estos extractos contienen pequeñas cantidades de algunos de los conservantes que se diluyen de manera temprana en el cromatograma. Por lo tanto, desviar la porción temprana del análisis minimizará la suciedad de la fuente MS. Establecer una ventana de tiempo de retención adecuada para cada analito. Esto debe basarse en mediciones de variación de tiempo de retención real para cada parámetro del método en las soluciones estándar CAL en la LC en el transcurso del tiempo. Un valor de más o menos tres veces la desviación estándar del tiempo de retención obtenido para cada analito mientras se establece la calibración inicial y se completa el IDC puede utilizarse para calcular un tamaño de ventana. Sin embargo, la experiencia del analista debe ser considerada de manera importante fuertemente para la determinación del tamaño de la ventana de retención adecuada. Establecer una calibración inicial válida o confirmar que la calibración es todavía válida ejecutando un CdC. Si se establece una calibración inicial, se debe completar el IDC. Comenzar analizando las muestras de campo, incluyendo muestras de control de calidad, en su frecuencia apropiada inyectando el mismo tamaño de alícuotas (en el desarrollo del método se utilizó 10 µL), bajo las mismas condiciones usadas para analizar los estándares de CAL. B.1.2.7.3 Análisis de datos y cálculos En la conclusión de adquisición de datos, usar el mismo software que se utilizó en el procedimiento de calibración para identificar los picos de interés en el tiempo de retención determinado. Utilizar el software del sistema de datos para examinar las concentraciones de iones de los picos en el cromatograma. Identificar un analito por comparación de su tiempo de retención con el del pico del analito de interés correspondiente en un estándar de referencia. El analista no debe extrapolar más allá del rango de calibración establecida. Si un área del pico del analito de interés excede el rango de la curva de calibración inicial, el extracto puede ser diluido con metanol que contiene 10% de agua. Volver a inyectar el extracto diluido. Incorporar el factor de dilución en los cálculos de concentración final. El rendimiento aceptable del SUR debe determinarse a partir del extracto de la muestra sin diluir. Los datos resultantes deben ser documentados como una dilución y el MRL debe ajustarse de acuerdo a ello. B.1.2.7.4 Informe de prueba Reportar la concentración de microcistina-LR en µg/L. B.1.2.8 Seguridad Cada reactivo utilizado en estos procedimientos debe ser tratado como un riesgo potencial para la salud y la exposición a estos materiales debe ser minimizada. Cada laboratorio es responsable de mantener un conocimiento de las regulaciones respecto a la manipulación segura de cualquier producto químico usado en este método. Debe ponerse a disposición de todo el personal involucrado en el análisis las hojas de datos de seguridad de los productos químicos. Algunas directrices de descontaminación/inactivación de toxinas pueden encontrarse en el libro "Biosafety in Microbiological and Biomedical Laboratories" y en otras referencias disponibles adicionales referentes a la seguridad del laboratorio. Los materiales estándar puros y las soluciones estándar de los analitos deben ser manejados con la protección adecuada para la piel y los ojos y se debe tener cuidado de no respirar los vapores o ingerir los materiales. B.1.2.9 Referencias · American Chemical Society Publication. "Safety in Academic Chemistry Laboratories," Committee on Chemical Safety, 7th Edition. Disponible en: · https://www.acs.org/content/dam/acsorg/about/governance/committees/chemicalsafety/publications/safety-in-academic-chemistry-laboratories-faculty.pdf (revisado el 17 de marzo de 2017). · EPA. 2015. METHOD 544. DETERMINATION OF MICROCYSTINS AND NODULARIN IN DRINKING WATER BY SOLID PHASE EXTRACTION AND LIQUID CHROMATOGRAPHY/TANDEM MASS SPECTROMETRY (LC/MS/MS) · Occupational Safety and Health Administration OSHA "OSHA Safety and Health Standards, General Industry". · U.S. Department of Health and Human Services, Public Health Service Centers for Disease Control and Prevention, National Institutes of Health. 2009. "Biosafety in Microbiological and Biomedical Laboratories", 5th edition, Appendix I Guidelines for Work with Toxins of Biological Origin. Disponible en https://www.cdc.gov/biosafety/publications/bmbl5/BMBL.pdf (revisado 17 de marzo de 2017). B.1.3 MÉTODO PARA LA DETERMINACIÓN DE MICROCISTINA MEDIANTE SPE Y LA CROMATOGRAFÍA DE LÍQUIDOS DE ALTO RENDIMIENTO (HPLC) CON DETECCIÓN (UV) ULTRAVIOLETA B.1.3.1 Símbolos y términos abreviados cm centímetro hPa hectoPascal HPLC cromatografía de líquidos de alta resolución kHz kilohertz MC microcistina MC-LR microcistina-LR MC-RR microcistina-RR MC-YR microcistina-YR min-1 por minuto NaClO hipoclorito sódico concentrado PDA arreglo de diodo psi libra por pulgada cuadrada SEC cromatografía de exclusión por tamaño SIM monitoreo selectivo de iones SPE extracción en fase sólida TFA ácido trifluoroacético UV ultravioleta B.1.3.2 Principio Las muestras de agua que contienen material o biomasa de cianobacterias deben ser filtradas primero. La biomasa se extrae por separado con un solvente (metanol/agua). El extracto es filtrado, diluido y se lleva a cabo una SPE para limpiar de muestra. El filtrado se trata como una muestra de agua pura. Las muestras de agua pura, así como muestras de agua de sistemas de abastecimiento se enriquecen usando SPE. Las microcistinas se eluyen de los cartuchos de SPE con una solución metanol/agua (90/10 volumen/volumen) que contiene 0.1% de TFA. La microcistina se cuantifica por cromatografía de líquidos de alta resolución, de fase reversa HPLC con detector ultravioleta y/o arreglo de diodos a 238 nm. B.1.3.3 Alcance y aplicación Este es un método para la determinación y cuantificación de microcistinas en muestras de agua colectadas a la entrada del sistema de tratamiento (que contiene biomasa) y de agua después del proceso de tratamiento, como es el agua en los sistemas de abastecimiento de agua de uso y consumo humano. El método descrito está validado para MC-RR, MC-YR y MC-LR. También es aplicable para la determinación de diversas variantes estructurales de las microcistinas, sin embargo, en estos casos, no puede realizarse una identificación inequívoca debido a la falta de estándares disponibles en el mercado y debido a la coelución. El valor de umbral de 1 µg/L de MC-LR en agua, puede ser seguido después del enriquecimiento con microcistina mediante SPE. B.1.3.4 Equipos y materiales En algunos casos se realizan referencias a marcas específicas y números de catálogo, sin embargo, estos se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden representar especificaciones adecuadas para los artículos. Algunos elementos de cristalería de laboratorio y equipo no se especifican, ya que su elección dependerá de las aplicaciones específicas y las circunstancias. Debe evitarse el uso de plásticos siempre que sea posible. Esto es necesario porque el uso de plásticos (por ejemplo pipetas de plástico, tubos de plástico o cartuchos de plástico) puede causar pérdidas de microcistinas por absorción en las paredes de la superficie. Agitador horizontal ajustable. Necesario sólo para el análisis de muestras que contiene fitoplancton. Baño ultrasónico Bomba binaria de HPLC. Adecuada para tasas de flujo de volumen entre 0.3 mL/min y 1 mL/min. Bomba de vacío para SPE Botellas de muestreo. Vidrio ámbar, estéril y previamente limpio. Cartuchos de SPE para el enriquecimiento de microcistina. La columna debe tener una capacidad mínima (cantidad de analito a ser retenido en la columna) no menor a 100 µg de cada microcistina y permitirá una recuperación no menor del 80% para MC-LR, así como no menor del 70% para el MC-RR y MC-YR cuando se aplica como una solución estándar en agua que contiene 0.05 µg de cada microcistina. La recuperación depende fuertemente de la marca y material del cartucho de SPE y de las especificaciones de los materiales como la carga de carbono, el tamaño de partícula etcétera. Los datos de recuperación se basan en cartuchos C-18 determinados por una sola medición. El cartucho debe tener las siguientes especificaciones de materiales: carga de carbón (16.9%), diámetro de la partícula (54 µm), cobertura de superficie (333 µg/m² basada en el %C) volumen de cartucho (3 ml), material por cartucho (500 mg). Si los valores de recuperación no pueden ser alcanzados, se recomienda cambiar la marca del cartucho SPE. Cartuchos de SPE de tipo disco pueden también utilizarse para el enriquecimiento de microcistina de muestras de agua. Centrífuga de laboratorio. 4000 min-1, fuerza centrífuga relativa (RCF) 10000g. Es recomendable el uso de una centrífuga a prueba de explosiones debido al uso de disolventes de extracción inflamables. Columna de HPLC. (p.ej. columna C18), empacada con material con un tamaño de partícula de 3 a 5µm; con un diámetro interior de 2 a 4.6 mm; y una longitud de 250 mm para asegurar la resolución de los estándares de referencia de MC-LR, MC-YR y MCRR. Debe utilizarse un protector de columna adecuado. El rango de presión debe ser de 70 000 hPa a 200 000 hPa (1 015 psi a 2 900 psi). Depósito de SPE. Capacidad de 500mL con conector para cartuchos. Detector de UV/arreglo de diodo (PDA). Con una longitud de onda =238 nm incluyendo corrección de fondo. El rango de longitud de onda de la PDA debe ser de 200 a 300 nm. El límite de detección (LOD) para el sistema debe ser 0.1 ng/µl (relación señal-ruido=3) y el límite de cuantificación (LOQ) debe ser 0.2 ng/µl (relación señal-ruido= 6) para cada microcistina (utilizando una solución estándar). Dispositivo de calefacción con control de temperatura y unidad de suministro de gas de nitrógeno. Con las siguientes características: Bloqueo de temperatura 30 a 50°C; temperatura del gas 20° C; pureza de gas 99.996%. Horno de columna HPLC. Con unidad de control de temperatura (35 °C). Papel de filtro de microfibra de vidrio. Tamaño de retención de 1 a 2 µm. El diámetro máximo del filtro debe ser de 47mm. La filtración es necesaria sólo para el análisis de las muestras que contienen fitoplancton. Sonda ultrasónica. Con características de 60W, 20 kHz Sistema de inyección. Con rango de volumen de inyección de 5 a 20µl. Unidad de filtro desechable. Tamaño de poro <0.45 µm Antes de usarlo, verifique a través de una prueba de recuperación que no existen pérdidas de microcistina durante la filtración. Existe la posibilidad de que diversos materiales puedan retener microcistinas. Opcionalmente puede utilizarse una microcentrífuga para evitar pérdidas. B.1.3.5 Reactivos y soluciones Use solamente reactivos de grado analítico reconocido y agua con grado 3, a menos que se especifique lo contrario. Acetonitrilo. CH3CN, grado HPLC Ácido de trifluoroacético. TFA, CF3COOH. Gradiente de fase móvil HPLC. Ejemplo en Tabla B.1.3-1, de este Apéndice. Tabla B.1.3-1. Gradiente de fase móvil de HPLC
Metanol. CH3OH, grado HPLC Microcistina. Comercialmente disponible en ampolletas. La calidad de la microcistina comercialmente disponible es muy variable, por lo que se recomienda realizar las soluciones de microcistina como se menciona en este documento. Solución de elución SPE. Metanol/agua (90/10 volumen/volumen) que contiene 0.1% de TFA. Solución estándar para dilución. Solvente de enjuague SPE y solvente de re-disolución. Metanol/agua (20/80 volumen/volumen). Solución de extracción. Metanol/agua (75/25 volumen/volumen). Solución de fase móvil HPLC (A). En un matraz volumétrico a 1 000 mL, añadir 800 mL de acetonitrilo y 500 µl de TFA y llevar al volumen final con acetonitrilo. Transferir esta solución en una botella de HPLC-eluyente. Desgasificar la solución antes de usar. Esta solución es estable a temperatura ambiente durante 3 semanas. Solución de fase móvil HPLC (B). En un matraz volumétrico de 1 000 mL, añadir 800 mL de agua y 500 µl de TFA y llevar al volumen final con agua. Transferir esta solución en una botella de HPLC-eluyente. Desgasificar la solución antes de usar. Esta solución es estable a temperatura ambiente durante 2 semanas. Solución de hidróxido de amonio. Solución de hidróxido de amonio NH4OH comercialmente disponible 1 mol/L. Se puede realizar la preparación de la solución partiendo de NH4OH concentrado. Solución de microcistina. Para determinar la concentración exacta de microcistina, para cada solución disolver en metanol puro la microcistina individual suministrada por el proveedor. Registrar la curva de absorción entre 220 y 250 nm en celdas de cuarzo de 1 cm en un espectrofotómetro con metanol en la celda de referencia. Calcular la concentración en masa de cada microcistina, i, en microgramos por mililitro, µg/mL, utilizando la ecuación: 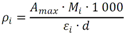 En donde: Amax absorbancia determinada en el máximo de la curva de absorción M masa molar de cada microcistina, en gramos por mol; g/mol absortividad molar de cada microcistina en metanol, en litros por (mol x centímetro); L/ (mol x cm) d longitud de camino óptico de la celda en centímetros, cm 1000 factor de cálculo para alcanzar la unidad de microgramos por mililitro, g/mL La M y la se listan en la Tabla B.1.3-2, de este Apéndice.
Para posteriores análisis HPLC, la proporción de solvente metanol/agua para los estándares de MC-LR, MC-YR y MC-RR se pueden ajustar a 20/80 volumen/volumen agregando el agua y permitiendo una concentración de 10 µg/ml para cada microcistina. Solución de tiosulfato de sodio. Disolver 1 g de tiosulfato de sodio Na2S2O3 (anhidro o con 5 H2O) en 100 mL de agua. La concentración final es =10/L (63 mmolar 63 en caso de Na2SO3 anhidro). Solución enriquecida para control del método. Preparar una solución enriquecida pipeteando 200 µl de la solución estándar mixta de microcistina en un matraz aforado de 500 mL. Diluir hasta la marca con agua (agua del sistema de abastecimiento o blanco de agua de fuente natural) y agitar bien. La concentración de esta salteada enriquecida es de 1 µg/L para MC-LR, MC-YR y MC-RR. Solución estándar mixta de microcistina. Pipetear los volúmenes de la solución de microcistina referidos en la Tabla B.1.3-3, de este Apéndice, en viales de 1 mL. Añadir a cada frasco, el volumen de la solución estándar para dilución referido en la Tabla B.1.3-3, de este Apéndice, para alcanzar un volumen final de 1000 µl y agitar bien. Tabla B.1.3-3. Esquema de pipeteo para las soluciones estándar mixta de microcistina
Solución mixta de microcistina. Preparar una solución estándar que contenga 2.5 µg/mL de cada microcistina (MC-LR, MC-YR, MC-RR) en la solución de dilución estándar. Almacenar por debajo de -16°C. Para evitar la incorporación de agua por la condensación, no abrir el frasco hasta que el contenido haya alcanzado la temperatura ambiente. Si la solución debe ser almacenada durante un largo periodo, use un frasco hermético. En caso de duda, pesar el frasco y registrar cualquier cambio en la masa durante el almacenamiento. B.1.3.6 Procedimiento B.1.3.6.1 Recolección, conservación y almacenamiento de muestras Recolectar las muestras de agua y almacenarlas no más de 48 horas en un lugar oscuro y fresco (4 a 8°C). B.1.3.6.2 Procedimiento analítico Antes del acondicionamiento, ajustar el cartucho SPE a temperatura ambiente. Para el acondicionamiento, deben de seguirse las especificaciones del fabricante. Sin embargo, si no se indican, pasar 4mL de metanol grado HPLC a través del cartucho. Posteriormente pasar 4 mL de agua a través del cartucho. Permitir un flujo de los solventes a una tasa <10 mL/min a través de la columna y asegurarse de que una pequeña porción del solvente permanezca en la cima de la columna hasta que se aplique la solución de la muestra. Para la preparación de la muestra, si esta se trata de agua del sistema de abastecimiento que ha pasado por un sistema de tratamiento, debe concentrarse la microcistina en las muestras de agua mediante la extracción en fase sólida. Si se trata de muestras de agua antes de entrar al tratamiento que puede contener fitoplancton, primero pasar la muestra (volumen recomendado: 50 a 100 mL) a través de un filtro (papel de filtro de microfibra de vidrio) para separar la biomasa de la fracción líquida. Si existen capas de algas flotantes un filtro puede ser insuficiente para la filtración de los 50 mL de agua. En este caso, reemplace inmediatamente el filtro tapado con uno nuevo. Concentrar la microcistina en las muestras de agua mediante la extracción en fase sólida. Extraer la biomasa en un filtro por separado seguido de la limpieza del extracto antes del análisis de HPLC. Si se usa un filtro gravimétrico, se puede determinar la masa de la biomasa y el contenido de microcistina puede ser mostrado en microgramos por gramo (µg/g). Para la extracción de microcistinas de las células en el filtro, deben extraerse las células en el filtro (si se usa más de un filtro, combinar los filtros), enjuagando el filtro(s) tres veces con 3 ml de la solución de extracción. Sonicar la solución en hielo durante 2 minutos con una sonda ultrasónica o en un baño ultrasónico. Después de eso, centrifugar la solución a 4000 min-1 durante 10 minutos a temperatura ambiente. Después de la centrifugación, reunir los sobrenadantes y secar 1mL de esta solución bajo una corriente de nitrógeno (40 °C). Antes de limpiar, disolver nuevamente los extractos en 500 µL de solución estándar de dilución y someter a ultrasonidos la muestra en un baño durante 5 minutos. Para el enriquecimiento de microcistina mediante la SPE, con el fin de evitar pérdidas, debe asegurarse que el pH de la muestra de agua está en el rango de 5.0 a 8.0. Si el pH está fuera de este rango, ajustar con ácido trifluoroacético o con solución de hidróxido de amonio, según corresponda. Añadir 500 µL de solución de tiosulfato de sodio a 500 mL de filtrado de agua de la muestra. Agitar bien y dejar reposar la mezcla durante 5 minutos. Añadir 5 mL de metanol grado HPLC y después de agitar, aplicar al cartucho acondicionado a una velocidad de flujo de 10 mL/min o menos (gotas visibles). Una vez que la muestra de agua ha pasado a través del cartucho, lavar el cartucho con 4mL de solución de dilución estándar. Eluir las microcistinas enriquecidas ya sea con 2.0 mL de solución de elución de SPE en un vial de vidrio de 4 mL (o siga las indicaciones del proveedor).Evaporar el eluyente hasta la sequedad con una corriente de nitrógeno (40 °C). Redisolver en 500 µL de solución estándar de dilución y someter a la muestra durante 5 min en un baño de ultrasonidos. Analizar este extracto directamente con HPLC. Para la limpieza de microcistinas mediante la SPE, aplicar las microcistinas extraídas aplican a la parte superior cartucho acondicionado y enjuagar el vial del extracto con 500 µL adicionales de solución estándar de dilución y aplicarlo también en la parte superior del cartucho. Después de que el líquido ha pasado a través del cartucho, enjuagar con 4 mL de solución de dilución estándar y desechar el eluído. Cuando la solución de enjuague haya pasado a través del cartucho, eluir las microcistinas limpias según las recomendaciones del proveedor, por ejemplo 2.0 mL de solución de elución de SPE en un vial de vidrio de 4 mL. Evaporar el eluyente hasta la sequedad con una corriente de nitrógeno (40°C), volver a disolver en 500 µL de solución estándar de dilución) y someter a ultrasonidos la muestra en un baño durante 5 minutos. Si la dilución de la muestra es necesaria, diluir 100 µL del extracto de muestra con 900 µL de solución estándar de dilución. Si la limpieza de cartuchos no reduce la coelución, pueden utilizarse como técnicas alternativas la cromatografía por exclusión de tamaño (SEC), la limpieza con columnas de inmunoafinidad o mediante el uso de materiales poliméricos. Para garantizar la máxima precisión durante la cromatografía de líquidos de alta resolución (HPLC), inyectar las microcistinas purificadas o enriquecidas según las instrucciones del fabricante en el puerto de inyección o válvula de inyección. Separar las microcistinas por HPLC a 35 °C con una columna de fase reversa utilizando el gradiente. Ajustar el flujo de volumen y el volumen de inyección según las dimensiones de la columna (diámetro interno, tamaño de partícula) para obtener la forma óptima de pico y resolución. Las microcistinas eluyen en el orden MC-RR, MC-YR y MC-LR y deben ser resueltas en línea base. Determinar los espectros de absorción entre 200 y 300 nm para confirmar la identificación, en la Fig. B.1.3-1 y Fig. B.1.3-2, de este Apéndice, se muestra respectivamente un cromatograma típico y los espectros de absorción típicos de microcistinas con detección de PDA. 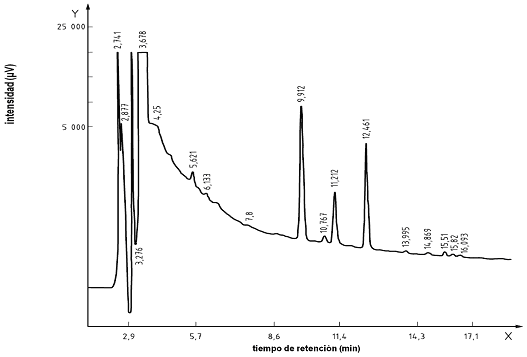 Fig. B.1.3-1 Cromatograma típico de MC-RR (9.91 min), MC-YR (11.21 min) y MC-LR (12.461 min) en una muestra de agua enriquecida después de SPE (concentración en la muestra de agua 1 000 ng/L) Condiciones de operación Volumen de inyección: 20 µl Columna: Phenomenex, LUNA, C18(2), 250 × 4.6 mm, 3 µm Tasa de flujo del volumen: 0.7 mL/min Fase móvil: Agua conteniendo 0.05%TFA; acetonitrilo conteniendo 0.05%TFA Detección: PDA a 238 nm 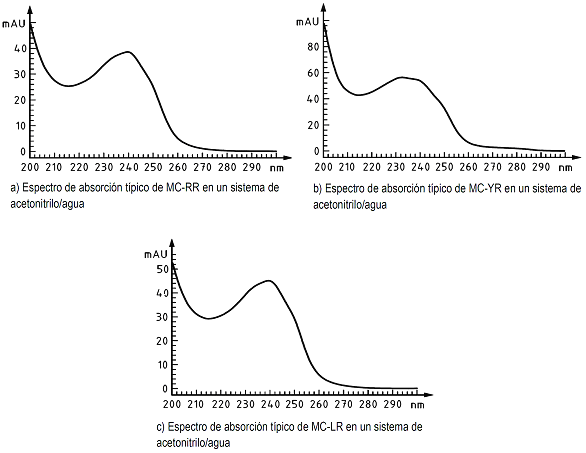 Fig. B.1.3-2 Espectros de absorción típica de MC-RR, MC-YR y MC-LR en un sistema de acetonitrilo/agua. Condiciones de operación Detección: HPLC-PDA Celda: Cuarzo B.1.3.6.3 Análisis de datos y cálculos Para calcular los resultados de las muestras de agua, se debe de calcular la concentración en masa de cada microcistina en microgramos por litro de la muestra según la siguiente ecuación, después de resolver la ecuación de la curva de calibración para cada microcistina: 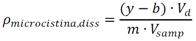 En donde: microcistina,diss concentración en masa de cada microcistina individual en microgramos por litro g/L Vd volumen de re-disolución de la muestra después de SPE en mililitros, mL Vsamp volumen de la muestra de agua aplicada en el cartucho SPE en litros, L y,b,m ver ecuación de curva de calibración Para calcular los resultados de biomasa, se debe calcular la concentración en masa de cada microcistina en fitoplancton en microgramos por mililitro de la muestra según la siguiente ecuación, después de resolver la ecuación de la curva de calibración para cada microcistina: 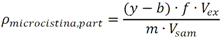 En donde: microcistina,dpart concentración en masa de cada microcistina individual en materia particulada en microgramos por mililitro, g/mL Vex volumen de extracción en mililitros, mL Vsam volumen de la muestra filtrada en mililitro, mL f factor de dilución en el paso de extracción de la célula; y,b,m ver ecuación de curva de calibración Como alternativa, puede utilizarse una cuantificación asistida por sistemas de cómputo. Reportar los resultados de las ecuaciones del punto B.1.3.6.3, de este Apéndice, por separado. Los resultados deben de ser sumados para muestras que contienen fitoplancton. Bajo condiciones naturales, la mayoría de las microcistinas están incluidas en el material particulado y generalmente menos del 20% está disuelta en el agua. Distintas microcistinas MC-RR, MC-YR y MC-LR pueden ser identificadas/reconocidas por sus espectros de absorción ultravioleta. En el caso de microcistinas diferentes a MC-LR, sus concentraciones en masa se pueden calcular utilizando la curva de calibración de MC-LR y los resultados se reportan como equivalentes MC-LR. B.1.3.6.4 Informe de prueba Reportar la concentración de microcistina-LR en µg/L. B.1.3.7 Calibración Determinar las señales cromatográficas a 238 nm (alturas de pico o áreas) de cada compuesto en la solución estándar mezclada de microcistina. Preparar las tres curvas de calibración (MC-RR, MC-YR y MC-LR) mediante la inyección de un volumen apropiado (5 a 20 µl) de las mezclas de microcistina descritas en los estándares de soluciones. Estas soluciones cubren el rango de 0.2 µg/ml a 3 µg/ml. Establecer la curva de calibración (método de mínimos cuadrados) para cada microcistina (área en el eje y y concentración en el eje x) utilizando una regresión lineal mostrada en la siguiente ecuación y verificando el trazo de linealidad. 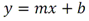 En donde: b punto de intercepción con el eje y (área); ordenada al origen m pendiente en mL por microgramos, ml/µg x valor del eje x en microgramos por mL, µg/mL y valor del eje y (área) El rango de trabajo se define como la parte lineal de la curva de calibración. Si el contenido de microcistina en las muestras se sitúa fuera del rango de calibración, ajustar el rango de calibración según las muestras. Alternativamente, puede diluirse la solución de inyección para el análisis HPLC con solución estándar de dilución a una concentración de microcistina apropiada para la curva de calibración establecida. Las diferencias en la concentración de metanol de la solución de inyección pueden tener un efecto en la cuantificación. Para la determinación de la recuperación, se debe efectuar el registro con la solución enriquecida. El nivel de picos debe estar dentro del rango de calibración (preferiblemente valores medios). B.1.3.8 Seguridad El método requiere el uso de soluciones que contienen microcistina. Las microcistinas son altamente hepatotóxicas para los seres humanos. Los residuos de microcistinas de laboratorio se recogerán por separado y se tratarán como residuos químicos altamente tóxicos. La descontaminación a largo plazo con hipoclorito sódico concentrado (NaClO) también es posible. Las personas que utilicen este método deben estar familiarizadas con las prácticas normales de laboratorio. Es responsabilidad del usuario establecer prácticas apropiadas de seguridad y salud y de cumplimiento de las condiciones reglamentarias. Es absolutamente esencial que los ensayos realizados de acuerdo con esta norma sean llevados por personal debidamente capacitado. B.1.3.9 Referencias · Blom, J.F.; Robinson, J.A.; y Jüttner, F. 2001. High grazer toxicity of [D-Asp3, (E)-Dhb7] microcystin- RR of Planktothrix rubescens as compared to different microcystins. Toxicon, 39, pp. 1923-1932 · International Organization for Standardization. 2005. Water quality- Determination of microcystins - Method using solid phase extraction (SPE) and high performance liquid Chromatography (HPLC) with ultraviolet (UV) detection. B.2 MÉTODO DE PRUEBA PARA LA DETERMINACIÓN DE QUISTES DE Giardia lamblia EN AGUA PARA USO Y CONSUMO HUMANO Giardia lamblia es un protozoario flagelado que se encuentra en las heces del hombre y animales más a menudo en la etapa de quistes, aunque en casos severos de diarrea acuosa se puede presentar la forma reproductiva o trofozoito. Este protozoario es el agente etiológico de la giardiasis y se ha observado que la ingestión de tan solo 10 quistes puede provocar la infección en humanos, sin embargo, es posible que el consumo de una cantidad menor de quistes viables en agua para beber sea suficiente para iniciar una infección. Los quistes de Giardia lamblia pueden sobrevivir en agua para uso y consumo humano por más de 2 meses a 8°C, por lo que la contaminación de las fuentes de abastecimiento de agua con materia fecal podría ocasionar brotes de giardiasis en la población expuesta a dicha agua. Los quistes de Giardia lamblia son más resistentes a la desinfección que las bacterias coliformes. La mayoría de los brotes asociados con fuentes de abastecimiento de agua y aguas de tipo recreacional han ocurrido como resultado de la ingestión de aguas superficiales que únicamente han sido cloradas, sin embargo, pueden ser removidos bajo procesos de potabilización adecuados. En los sistemas de abastecimiento de agua, la concentración de quistes de Giardia lamblia es relativamente baja, por lo que el análisis de muestras de agua de uso y consumo humano requiere métodos de muestreo que incluyan la concentración de quistes de Giardia lamblia a partir de grandes volúmenes (L) de agua filtrando a través de arena, membranas o filtros microporosos profundos. De manera posterior a la toma de muestra, es necesario llevar a cabo la purificación de los quistes que se encuentran en la muestra y que se han sedimentado, esto generalmente a través de técnicas de flotación utilizando soluciones de sulfato de zinc, sacarosa, citrato de potasio o Percoll-sacarosa. A partir de los quistes purificados puede realizarse la determinación a través de observación microscópica usando cromógenos o técnicas de tinción con anticuerpos fluorescentes y criterios morfológicos. Por lo anterior, para fines del cumplimiento de esta Norma a continuación se describen los siguientes métodos: · Método de muestreo para la determinación de quistes de Giardia lamblia en muestras de agua para uso y consumo humano (contenido en el punto B.2.1, de este Apéndice). · Método de purificación de quistes de Giardia lamblia en muestras de agua para uso y consumo humano (contenido en el punto B.2.2, de este Apéndice). · Método microbiológico para la determinación de quistes de Giardia lamblia en muestras de agua para uso y consumo humano (contenido en el punto B.2.3, de este Apéndice). Los avances en la investigación y la atención de sospechas de brotes de enfermedades transmitidas por agua han permitido el desarrollo de diversos métodos de prueba para la determinación de quistes de Giardia lamblia en agua para uso y consumo humano. Estos métodos evolucionan rápidamente, por lo que para el cumplimiento de esta Norma para la determinación de quistes de Giardia lamblia en muestras de agua para uso y consumo humano se podrán utilizar de manera indistinta, el método microbiológico descrito en el punto B.2.3, de este Apéndice y las técnicas de inmunofluorescencia (contenido en el punto B.2.4, de este Apéndice) o de biología molecular (contenido en el punto B.2.5, de este Apéndice) descritas en este método de prueba. B.2.1 MÉTODO DE MUESTREO PARA LA DETERMINACIÓN DE QUISTES DE Giardia lamblia EN MUESTRAS DE AGUA PARA USO Y CONSUMO HUMANO B.2.1.1 Principio El presente método incluye la toma de muestra en campo después del proceso de potabilización. La toma de muestra en sitio incluye la concentración de los quistes a través de un cartucho filtrante partiendo de un volumen de agua suficiente para la determinación de quistes de Giardia lamblia, así como la preservación de la muestra hasta su entrega al laboratorio. B.2.1.2 Alcance y aplicación Este método describe el procedimiento de muestreo-concentración en campo de agua después del proceso de potabilización y la preservación de la muestra hasta la entrega al laboratorio del cartucho filtrante para la determinación de quistes de Giardia lamblia presentes en muestras de agua para uso y consumo humano. B.2.1.3 Equipos y materiales Las referencias a marcas específicas y números de catálogo se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden representar especificaciones adecuadas para los artículos. Bolsas para muestreo de cierre hermético de 1 600 a 3 000 mL. Conexión hembra para manguera con rosca. Conexión macho para manguera con rosca o conexiones de ensamble rápido macho y hembra, para el módulo de entrada (los diámetros de las conexiones y el tipo de rosca dependerán de las medidas del portacartucho que utilice). Hielera de tamaño adecuado para la cantidad de muestras a transportar. Llave para limitar el flujo de salida Manguera de jardín, transparente (longitud suficiente para realizar las conexiones de entrada y descarga). Material filtrante. Puede ser un cartucho filtrante de hilo encordado (de fibras acrílicas, de polipropileno u otro material disponible que no libere fibras) de 25 cm de longitud y una porosidad de 1 µm o pueden utilizarse membranas de filtración con poro de 1 µm. El uso de un cartucho filtrante de hilo o de una membrana de filtración dependerá del dispositivo de muestreo que se ensamble. Medidor de flujo Motobomba de gasolina o eléctrica, en caso de que la muestra deba colectarse de tanques, estanques de almacenamiento o desde flujos después de la potabilización que no permitan la toma de muestra mediante una conexión o dispositivo de muestreo en la planta de potabilización, o bien, cuando la presión en la línea sea menor a 100 kilopascales (kPa). La motobomba debe ser instalada al final del módulo de salida para evitar el riesgo de contaminación cruzada de muestras previamente filtradas. Porta cartucho de polietileno, polipropileno, policarbonato u otro material disponible, capaz de contener un filtro cartucho de 25 cm de longitud. B.2.1.4 Procedimiento B.2.1.4.1 Procedimiento de muestreo Ensamblar el dispositivo de muestreo como se muestra en la Fig. B.2.1-1a. En caso de que se cuente con una conexión para muestreo a la salida de la planta de potabilización, conectar a ella el dispositivo (Fig. B.2.1-1a, de este Apéndice). En caso de que no se cuente con una conexión para muestreo a la salida de la planta de potabilización, introducir la manguera en el cuerpo de agua posterior a la potabilización de donde se va a tomar la muestra y conectar la motobomba a la salida del dispositivo después del módulo de descarga (Fig.B.2.1-1b, de este Apéndice). 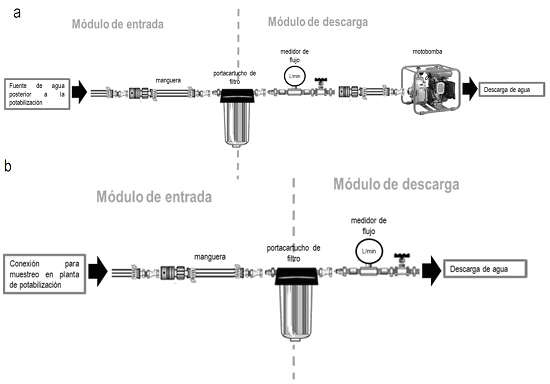 Fig. B.2.1-1 Dispositivo de muestreo. 1a) en caso de existir conexión para muestreo a la salida de la planta de potabilización. 1b) en caso de no existir conexión para muestreo a la salida de la planta de potabilización. Permitir el flujo del agua a través del dispositivo de muestreo abriendo la llave de la conexión para muestreo o en su caso encender la motobomba. Regular el flujo a una tasa aproximada de 3.8 L/min con la llave de la conexión para muestreo de la planta de potabilización y regulando la potencia de la motobomba. La dirección del flujo del agua debe ser desde la superficie externa del filtro hacia el interior del mismo. Registrar el tiempo y la lectura del medidor de flujo al inicio. Permitir el flujo de un volumen aproximado de 380 L de agua. Detener el flujo del agua en el dispositivo de muestreo cerrando la llave de la conexión para muestreo o en su caso apagando la motobomba. Registrar el tiempo y la lectura del medidor de flujo al final. Desconectar el dispositivo de la conexión para muestreo procurando mantener el extremo abierto de la conexión de entrada por encima del nivel del extremo abierto de la conexión de salida para prevenir posibles pérdidas de material particulado por retro lavado del filtro. Una vez desconectado, drenar el agua que quedo en el dispositivo tanto como sea posible. B.2.1.4.2 Preservación y transporte de la muestra El cartucho filtrante contenido en el portacartucho puede ser llevado al laboratorio para su análisis dentro del portacartucho o fuera de él. Para transportar el cartucho filtrante dentro del portacartucho, obstruir las conexiones de entrada y salida del portacartucho con el filtro dentro, etiquetando el portacartucho, en este caso no debe drenar el agua restante ya que será incorporada en los lavados posteriores durante la extracción del filtro en el laboratorio. Para transportar el cartucho fuera del portacartuchos, abrir el dispositivo y asépticamente retirar el cartucho filtrante y colocarlo en una bolsa de plástico etiquetada, cerrar la bolsa, introducirla en una segunda bolsa y cerrar esta última bolsa. En ambos casos, tanto el cartucho filtrante dentro del portacartuchos como el cartucho filtrante fuera del mismo, deberán ser refrigerados en la hielera con hielo o con geles congelantes tan pronto como sea posible después de realizado el muestreo. En ningún caso debe congelarse la muestra y debe ser transportada al laboratorio de análisis para su procesamiento tan pronto como sea posible sin exceder de las 48 horas después de su colecta. Debe procurarse reducir al mínimo los tiempos de transporte y almacenamiento. B.2.2 MÉTODO DE PURIFICACIÓN DE QUISTES DE Giardia lamblia EN MUESTRAS DE AGUA PARA USO Y CONSUMO HUMANO B.2.2.1 Símbolos y términos abreviados g gramos Tween 80 monooleato de polioxietilen-sorbitano v/v volumen/volumen B.2.2.2 Principio Los quistes de Giardia lamblia concentrados a partir de una muestra de agua de gran volumen, y que se encuentran retenidos en el cartucho filtrante, deben ser extraídos para ser purificados; eliminando sedimento, detritus y demás materiales que interfieran con la identificación, determinación y cuantificación. La purificación de los quistes se realiza generalmente a través de técnicas de flotación utilizando soluciones de sulfato de zinc (ZnSO4), sacarosa, citrato de potasio o Percoll-sacarosa. B.2.2.3 Alcance y aplicación Este método es utilizado en laboratorio para la extracción y purificación de los quistes de Giardia lamblia retenidos en el material filtrante a partir del flujo de una muestra de gran volumen de agua para uso y consumo humano durante el procedimiento de muestreo descrito en el punto B.2.1, de este Apéndice. B.2.2.4 Equipos y materiales Las referencias a marcas específicas y números de catálogo se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden representar especificaciones adecuadas para los artículos. Botellas para centrífuga Centrífuga capaz de alcanzar al menos 660 g y que permita procesar volúmenes de 3 a 4 L Charola de Aluminio o acero inoxidable de 50 x 35 cm aprox. o papel aluminio Guantes de nitrilo o látex Mango para bisturí o una navaja afilada que permita un manejo aséptico Micropipeta de volumen variable de 5-100 µL Navajas para bisturí del tamaño adecuado Puntas para micropipeta de 5-50 µL, 100 µL. Sifón Sistema de vacío con trampa (Matraz Kitasato de 4L; tapón de silicón; tubo de vidrio de 6mm de diámetro u otro adecuado; mangueras de plástico flexible; punta para micropipeta de 500-100 µL) Tubos de centrífuga de 15 y 50 mL Vaso de precipitados de polipropileno de 4 L Vórtex B.2.2.5 Reactivos y soluciones Agua destilada grado reactivo Formaldehído al 37% (v/v) Disolución de formaldehído al 2% (v/v) Tween 80 al 0.1% B.2.2.6 Procedimiento B.2.2.6.1 Liberación de quistes Los quistes retenidos en el material del cartucho filtrante o en la membrana de filtración deben de ser extraídos antes del proceso de purificación para eliminar detritus y otros materiales retenidos en el cartucho filtrante que interfieran con la determinación de los quistes de Giardia lamblia. En caso de que se entregue al laboratorio el portacartucho con el cartucho filtrante o membrana de filtración en su interior, deberá abrir el portacartuchos y retirar el material filtrante (cartucho filtrante o membrana de filtración) del portacartuchos vaciando el agua contenida en el portacartuchos dentro de un vaso de precipitados de 4 L. En caso de que se entregue al laboratorio el cartucho filtrante o la membrana de filtración dentro de bolsas de muestreo, deberá abrir la bolsa secundaria y primaria para retirar el cartucho filtrante o la membrana de filtración. Las membranas o filtros utilizados deberán ser manipulados en condiciones asépticas utilizando guantes de nitrilo o látex. En el caso de que se trate de cartuchos filtrantes, colocarlo sobre una charola de aluminio o acero inoxidable o sobre una hoja de papel aluminio. Con la ayuda de un bisturí que permita un manejo aséptico, realizar cortes longitudinales a todo lo largo del cartucho y separar las fibras del filtro de la estructura central del cartucho. Cortar las fibras en dos o más porciones aproximadamente iguales (incluir tanto la parte externa como la parte interna del filtro) y separar las fibras de cada porción tanto como sea posible. Es frecuente que se observe una clara diferencia entre la parte interna y externa de los filtros de acuerdo a la profundidad a la cual ha penetrado el sedimento en el cartucho del filtro. Alternativamente, localizar el final del hilo en el exterior del cartucho, desenrollar las fibras y dividir en porciones aproximadamente iguales. Lavar cada porción por separado en vasos de precipitados de polipropileno de 4 L, con 1 L de agua destilada grado reactivo o con una disolución de Tween 80 al 0.1%. La muestra se agita y se presiona repetidamente durante 10 minutos procurando extraer todas las partículas que pudieran estar atrapadas entre las fibras. Para separar con más eficacia el material particulado, deberá amasar las fibras manualmente o agitarlas manual o mecánicamente durante 10 a 15 minutos, en este último caso agregando el volumen suficiente para cubrir las fibras. Exprimir cada porción y recoger todo el fluido combinándolo todo en un solo vaso, en su caso, en el mismo vaso de precipitados con el agua que estaba contenida en el portacartuchos. Si las fibras aun retienen cantidades significativas de material particulado repetir el proceso de extracción hasta que las fibras aparezcan limpias. En caso de que se trate de membranas de filtración, deberá retirar la membrana del dispositivo de filtración y colocarla en 1 L de agua destilada grado reactivo o de una disolución de Tween 80 al 0.1%. La muestra se agita vigorosamente durante 10 minutos procurando extraer todas las partículas atrapadas en la superficie de la membrana. El extracto obtenido puede ser sometido al procedimiento de concentración de quistes. Sin embargo, en caso de no poder continuar con el procedimiento el mismo día este puede preservar adicionando un volumen suficiente de formaldehido al 37% (v/v) para tener una concentración final de 2% (v/v). Refrigere este extracto preservado en un vaso de precipitados de 4 L o en botellas para centrifuga hasta continuar con el proceso de concentración al día siguiente. B.2.2.6.2 Concentración de quistes La refrigeración durante una noche, tanto en el vaso de precipitados como en las botellas para centrifuga, permitirá la sedimentación del extracto. Si el extracto se refrigeró y sedimentó en el vaso de precipitados de 4 L durante toda la noche, deberá decantar o aspirar el sobrenadante con un sifón o un sistema de vacío con trampa. Transferir el sedimento a un tubo de centrifuga del volumen apropiado (50 mL preferiblemente) y resuspenderlo en un volumen de formaldehido al 2% equivalente al volumen total del sedimento, en caso de que el sedimento sea escaso, resuspender en 10 mL de la disolución (utilice la mitad del volumen para resuspender y vaciar al tubo de centrífuga, y la mitad restante para lavar las paredes del recipiente donde se encontraba el sedimento). Puede ser necesario ocupar un poco más de disolución de formaldehido para los lavados. El extracto resultante está listo para ser centrifugado y continuar con el proceso de concentración de los quistes. Si el extracto se refrigeró y sedimentó en botellas de centrifuga, estimar el volumen de los sedimentos y concentrar todos los sedimentos en un solo tubo de centrifuga (50 mL preferiblemente). De ser necesario utilizar un poco del volumen estimado de formaldehido al 2% para resuspender los sedimentos y vaciarlos y el resto para lavar las paredes de las botellas de centrífuga. Puede ser necesario ocupar un poco más de disolución de formaldehido para los lavados. El extracto resultante está listo para ser centrifugado y continuar con el proceso de concentración de los quistes. Si el extracto no fue refrigerado ni sedimentado y es sometido al procedimiento de concentración de los quistes, está listo para ser centrifugado y continuar con el proceso de concentración de los quistes. Centrifugar el extracto obtenido a 660 g durante 5 minutos. Decantar o aspirar el sobrenadante con ayuda de una micropipeta tanto como sea posible sin alterar o resuspender el sedimento. Resuspender el sedimento en una disolución de formaldehído al 2%, transferirlo a un tubo para centrífuga de 15 mL y agitar con un vórtex. La cantidad de formaldehido adicionado deberá ser igual a la cantidad del sedimento obtenido. Centrifugar a 660g durante 5 minutos. Decantar o aspirar el sobrenadante con ayuda de una micropipeta tanto como sea posible sin alterar o resuspender el sedimento. Si el sedimento obtenido es escaso, resuspender en 10 mL de la disolución de formaldehído al 2%, agitar con un vórtex y centrifugar a 600 g durante 5 minutos. Si el volumen del sedimento es menor o igual a 1 mL, decantar o aspirar el sobrenadante en su totalidad y desechar el sobrenadante. Si el volumen del sedimento es mayor a 1 mL resuspender en el sobrenadante y transferir volúmenes equivalentes de 1mL en tubos de centrifuga de 15 mL de capacidad, volver a centrifugar por 5 minutos a 660 g. Decantar y desechar el sobrenadante. El volumen de sedimento restante en cada tupo debe ser de aproximadamente 1 a 2 mL. B.2.3 MÉTODO MICROBIOLÓGICO PARA LA DETERMINACIÓN DE QUISTES DE Giardia lamblia EN MUESTRAS DE AGUA PARA USO Y CONSUMO HUMANO B.2.3.1 Símbolos y términos abreviados ZnSO4 sulfato de zinc B.2.3.2 Principio A partir de los quistes purificados puede realizarse la determinación de estos a través de observación microscópica usando cromógenos o técnicas de tinción con anticuerpos fluorescentes y criterios morfológicos. B.2.3.3 Alcance y aplicación Este método es utilizado para la determinación de quistes de Giardia lamblia, purificados a través del método descrito en el punto B.2.2, de este Apéndice, presentes en muestras de agua para uso y consumo humano colectadas al final del sistema de potabilización, a través de un método microbiológico por microscopia óptica con tinción con lugol. De manera adicional a este método microbiológico por microscopia óptica con tinción con lugol, para la determinación de quistes de Giardia lamblia pueden utilizarse las técnicas de inmunofluorescencia (punto B.2.4, de este Apéndice) o de biología molecular (punto B.2.5, de este Apéndice) descritas en este método de prueba. B.2.3.4 Equipos y materiales Las referencias a marcas específicas y números de catálogo se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden representar especificaciones adecuadas para los artículos. Aceite de inmersión Asa bacteriológica estéril Centrífuga capaz de alcanzar al menos 660 g y que permita procesar volúmenes de 3 a 4 L Cubreobjetos de 24 x 50 mm, limpios y libres de grasa Frascos con gotero de volumen adecuado Micropipeta de volumen variable de 5-100 µL Microscopio óptico de campo claro. Palillo o aplicador de madera estéril Portaobjetos de 25 x 75 mm, limpios y libres de grasa Puntas para micropipeta de 5-50 µL, 100 µL. Tubos de centrífuga de 15 y 50 mL Vórtex B.2.3.5 Reactivos y soluciones Disolución de Lugol, la disolución madre consiste en 20 g de yodo metálico y 40 g de yoduro potásico, disueltos en un litro de agua destilada. Para preparar la disolución de trabajo diluya la disolución madre 1:5 con una disolución de ZnSO4 sulfato de zinc con una densidad de 1.20 (gramos por mililitro). Disolución de Sulfato de Zinc con una densidad de 1.20 (gramos por mililitro) preparada a partir de ZnSO4 Sulfato de Zinc hepta hidratado, grado reactivo analítico Vaselina sólida o algún sellador para muestras biológicas en portaobjetos B.2.3.6 Procedimiento B.2.3.6.1 Montaje de laminillas Tomar el volumen de a 2 mL de sedimento resultante durante la concentración de los quistes realizada en el procedimiento de purificación (punto B.2.2, de este Apéndice). Adicionar de 1 a 3 gotas de la disolución de trabajo de lugol 1:5 al sedimento y homogenizar manualmente con un palillo de madera estéril. Agregar 5mL de la disolución de ZnSO4 de densidad 1.20 (gramos por mililitro), si utilizó un aplicador de madera, introducirlo en el tubo con el sedimento y el lugol al momento de agregar la disolución de sulfato de zinc (ZnSO4) para recuperar posibles pérdidas. Mezclar de manera manual o con ayuda de un vortex y dejar reposar por 5 minutos. Continuar adicionando la disolución de sulfato de zinc (ZnSO4) hasta la capacidad máxima del tubo, teniendo cuidado de evitar derrames. Centrifugar por 3 minutos a 650 g. Abrir la tapa de la centrifuga con cuidado y sin tocar ni agitar los tubos dejar reposar de 2 a 5 minutos. Los quistes de Giardia lamblia que se encontraban contenidos en la muestra de agua se encontrarán suspendidos en el sobrenadante formando, en la mayoría de los casos, una película flotante en la superficie del mismo. Con la ayuda de un asa estéril recoger la película flotante en la superficie y colocarla en un portaobjetos rotulado, esta operación se puede repetir 2 o 3 veces para asegurar una mayor recuperación. Es necesario hacer laminillas hasta agotar la película flotante. El quiste está en la superficie no en el sedimento. Colocar un cubreobjetos de 24 x 50 mm y selle la muestra con vaselina sólida o con algún otro sellador disponible. Los cubreobjetos y portaobjetos deberán manipularse cuidadosamente y por los bordes. B.2.3.6.2 Observación microscópica Las laminillas montadas en el apartado anterior, deben observarse bajo el microscopio óptico con el objetivo de 100X con aceite de inmersión, realizando un barrido total del cubreobjetos como se muestra en la Fig. B.2.3.-1, de este Apéndice.  Fig. B.2.3-1 Barrido de la muestra La determinación de los quistes de Giardia lamblia deberá ser realizada por personal capacitado en esta actividad, teniendo cuidado de no confundir los quistes de Giardia lamblia con otros microorganismos como levaduras, diatomeas, Coccidias u otros organismos y siguiendo los siguientes criterios de inclusión: · nitidez (pared del quiste bien definida) · forma ovalada · tamaño de 8 a 18µm de longitud · estructura ovalada · pared gruesa llamada pared quística que mide 0.3 a 0.5µm · presencia de por lo menos dos de las siguientes características internas: presencia de núcleos (generalmente son cuatro y siempre aparecen dispuestos en alguno de los polos), cuerpos basales y axonemas. El uso de microfotografías de quistes de Giardia lamblia obtenidas en referencias bibliográficas pueden ser de gran utilidad para la determinación de estos. B.2.3.6.3 Análisis de datos Registrar las siguientes características: · tamaño y forma del quiste · presencia de núcleos · presencia de cuerpos parabasales · presencia de axonemas Registrar el número de quistes de Giardia lamblia determinados en el total del sedimento, correspondiente al volumen de agua pasado a través del material filtrante (380 L) de acuerdo con el procedimiento de muestreo descrito (punto B.2.1, de este Apéndice). B.2.3.7 Informe de prueba Reportar ausencia o presencia de quistes de Giardia lamblia en el volumen total de la muestra filtrada. B.2.3.8 Referencias Standard Methods for the Examination of Water and Wastewater. 9711.B Giardia lamblia. 18a Ed.1992. B.2.4 TÉCNICAS DE INMUNOFLUORESCENCIA PARA LA DETERMINACIÓN DE QUISTES DE Giardia lamblia EN MUESTRAS FILTRADAS DE AGUA PARA USO Y CONSUMO HUMANO De manera adicional al método microbiológico por microscopia óptica con tinción con lugol, para la determinación de quistes de Giardia lamblia pueden utilizarse las técnicas de inmunofluorescencia. Las técnicas de inmunofluorescencia para la determinación de quistes de Giardia lamblia evolucionan rápidamente debido a los avances científicos y tecnológicos, por lo cual no se establece un método específico para la determinación de quistes de Giardia lamblia en el presente apartado. Sin embargo, el usuario que utilice técnicas de inmunofluorescencia para la determinación de quistes de Giardia lamblia en muestras filtradas de agua y consumo humano posterior a la potabilización para el cumplimiento de esta Norma, deberá consultar la literatura actual y especializada para las técnicas y metodologías más recientes. Entre los métodos que pueden ser utilizados son los siguientes: · EPA. 1995. ICR Protozoan Method for Detecting Giardia Cysts and Cryptosporidium Oocysts in Water by a Fluorescent Antibody Procedure · EPA. 2005. Method 1623: Cryptosporidium and Giardia in Water by filtration/IMS/FA La aplicación de técnicas de inmunofluorescencia es a menudo realizada a partir de kits disponibles comercialmente por ejemplo, entre otros, el kit EasyStainTM /BTF- A Biomerieux Company, por lo cual el usuario que utilice kits disponibles comercialmente para realizar técnicas de Inmunofluorescencia para la determinación de quistes de Giardia lamblia en muestras de agua y consumo humano posterior a la potabilización para el cumplimiento de esta Norma, debe de asegurarse que el kit utilizado incluya cuando menos: · Reactivo de inmunofluorescencia altamente específico para detectar la presencia de quistes de Giardia lamblia en muestras de agua · Anticuerpos monoclonales IgGI altamente purificados para evitar reacciones cruzadas · Control positivo en una concentración aproximada de 200 a 400 quistes por µl · Aprobación de organismos internacionales como United States Environmental Protection Agency (Method EPA 1622/1623), United Kingdom Drinking Water Inspectorate o Japanese Water work Association Las referencias a marcas específicas se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden únicamente representar especificaciones adecuadas para los artículos. B.2.5 TÉCNICAS DE BIOLOGÍA MOLECULAR PARA LA DETERMINACIÓN DE QUISTES DE Giardia lamblia EN MUESTRAS FILTRADAS DE AGUA PARA USO Y CONSUMO HUMANO De manera adicional al método microbiológico por microscopia óptica con tinción con lugol, para la determinación de quistes de Giardia lamblia pueden utilizarse las técnicas de biología molecular. Las técnicas de biología molecular para la determinación de quistes de Giardia lamblia evolucionan rápidamente debido a los avances científicos y tecnológicos, por lo cual no se establece un método específico para la determinación de quistes de Giardia lamblia en el presente punto. Sin embargo, el usuario que utilice técnicas de biología molecular para la determinación de quistes de Giardia lamblia en muestras filtradas de agua y consumo humano posterior a la potabilización para el cumplimiento de esta Norma, deberá consultar la literatura actual y especializada para las técnicas y metodologías más recientes. La aplicación de técnicas de biología molecular es a menudo realizada a partir de kits disponibles comercialmente por ejemplo, por lo cual para la determinación de quistes de Giardia lamblia en muestras filtradas de agua y consumo humano posterior a la potabilización por técnicas de biología molecular para el cumplimiento de esta Norma, se podrán utilizar kits de biología molecular como Kit TaqMan de reacción en cadena de la polimerasa (PCR) para Giardia lamblia, Kit Verde Fluorescente "SYBR Green" PCR para Giardia lamblia o Kit PCR punto final para Giardia lamblia o cualquier otro comercialmente disponible. Las referencias a marcas específicas se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes, los cuales se recomienda que incluyan lo siguiente: En el caso de kits TaqMan PCR para Giardia lamblia: · Desoxinucleótidos trifosfatados (deoxyNTPs), Cloruro de magnesio (MgCl2), inhibidor de ribonucleasas (RNAsas), agua libre de RNAsas, polimerasa, amortiguador, fluoróforo pasivo o mezcla maestra (Master mix) · control para la detección PCR, para monitorear la inhibición de la reacción y validar la calidad del proceso · cebador de ácido desoxirribonucléido (DNA) (primer) o mezcla de Sondas (Probe mix) para la amplificación de una secuencia específica del genoma de Giardia lamblia · control positivo y control negativo, para confirmar la integridad de los reactivos del kit En el caso de kits SYBR Green qPCR para Giardia lamblia: · deoxyNTPs, MgCl2, inhibidor de RNAsas, agua libre de RNAsas, polimerasa, amortiguador, fluoróforo pasivo o mezcla maestra (Master mix) · cebadores de DNA (primers) para la amplificación de una secuencia específica del genoma de Giardia lamblia · control positivo y control negativo, para confirmar la integridad de los reactivos del kit y un control interno para la validación de PCR En el caso de kits PCR punto final para Giardia lamblia: · deoxyNTPs, MgCl2, inhibidor de RNAsas, agua libre de RNAsas, polimerasa, amortiguador o mezcla maestra (Master mix) · cebadores de DNA (primers) para la amplificación de una secuencia específica del genoma de Giardia lamblia · control positivo y control negativo para confirmar la integridad de los reactivos del kit · materiales necesarios para visualizar los segmentos amplificados del genoma de Giardia lamblia por electroforesis (buffer de carga, marcador de peso molecular, etc.) Las referencias a marcas específicas se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden únicamente representar especificaciones adecuadas para los artículos. B.3 MÉTODO PARA LA DETERMINACIÓN DE BTEX (BENCENO, TOLUENO, ETILBENCENO Y XILENOS) Y ESTIRENO EN AGUA PARA USO Y CONSUMO HUMANO POR CROMATOGRAFÍA DE GASES CON DETECTOR DE ESPECTROMETRÍA DE MASAS B.3.1 Símbolos y términos abreviados BFB 4-bromofluorobenceno BTEX benceno, tolueno, etilbenceno y xilenos (orto, meta y para) CdC curva de calibración CG cromatógrafo de gases CG/EM cromatografía de gases con detector de espectrometría de masas DE desviación estándar EICP perfil común de iones extraídos EM espectrometría de masas EMIE ionización de impacto electrónico FR factor de respuesta FRR factores de respuesta m/z masa/carga PCIE perfil de corriente de iones extraídos PTFE politetrafluoroetileno R coeficiente de correlación adimensional %DER desviación estándar relativa B.3.2 Principio Los compuestos orgánicos volátiles son transferidos eficientemente de la fase acuosa a la fase gaseosa mediante burbujeo de un gas inerte en la muestra. La fase gaseosa es arrastrada a través de una trampa en la cual se adsorben los analitos de interés. Al finalizar la purga, la trampa se calienta para desorber los compuestos y con el mismo gas inerte son introducidos en la columna cromatográfica. El cromatógrafo de gases se programa a una temperatura para separar los analitos de la muestra, los cuales son identificados y a su vez cuantificados por el espectrómetro de masas. La cuantificación se realiza comparando la respuesta de los iones característicos con una curva de calibración. La identificación de los analitos de interés se logra comparando sus espectros de masas con los espectros de masas de estándares. La cuantificación se logra comparando la respuesta de un ion (cuantificación) con respecto a un estándar interno usando una curva de calibración apropiada para la aplicación deseada. B.3.3 Alcance y aplicación El método descrito es un procedimiento para la determinación de compuestos orgánicos volátiles, específicamente para benceno, tolueno, etilbenceno y xilenos (BTEX) así como estireno en muestras de agua de uso y consumo humano através de cromatografía de gases con detector de espectrometría de masas (CG/EM). Existen varias técnicas mediante las cuales estos compuestos pueden introducirse en el sistema CG/EM, la técnica más común para analitos orgánicos volátiles es el método de purga y trampa. B.3.4 Equipos y materiales Las referencias a marcas específicas y números de catálogo se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden representar especificaciones adecuadas para los artículos. Balanza analítica. Con sensibilidad de 0.1 mg. Cámara de purga. Diseñada para aceptar muestras de 5 o 25 mL con una columna de agua de al menos 3 cm de profundidad. El espacio libre superior (headspace) gaseoso entre la columna de agua y la trampa debe tener un volumen total de al menos de 5 mL. El gas de purga debe pasar a través de la columna de agua en forma de burbujas finamente divididas con un diámetro menor de 3 mm. Introducir el gas de purga no más de 5 mm de la base de la columna de agua. El dispositivo de purga ilustrado en la Fig. B.3-1, de este Apéndice, cumple estos criterios. Se pueden utilizar otros tipos de dispositivos de purga siempre y cuando se demuestre que su desempeño es adecuado. 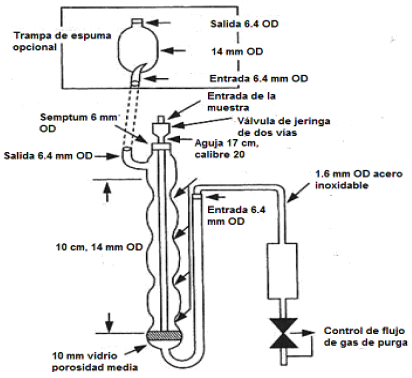 Fig. B.3-1 Dispositivo de purga. Se incluye sólo como ejemplo y no implican aprobación de los productos. Pretende únicamente representar especificaciones adecuadas para los artículos. Columnas capilares: usar cualquier columna capilar de CG que cumpla con los criterios de desempeño. Asegurarse que el flujo del desorbente sea compatible con la columna elegida. Abajo se enlistan cuatro ejemplos de columnas aceptables. Columna 1: de 60 m de longitud x 0.25 mm ID VOCOL de amplio calibre, con 1.5 µm de espesor de película. Columna 2: de 30 m de longitud x 0.53 mm, con 3 µm de espesor de película. Columna 3: de 30 m de longitud x 0.32 mm, con 1 µm de espesor de película. Columna 4: de 30 m de longitud x 0.25 mm, con 1.4 µm de espesor de película. Cromatógrafo de gases (CG) con detector de espectrometría de masas (EM). Utilizar un CG con temperatura programable adecuado para la inyección sin división (splitless), con división (Split) y con controladores de flujo. Utilizar un espectrómetro de masas (EM), capaz de escanear de 35 a 270 uma (unidad de masa atómica) cada segundo o menos, utilizando 70 eV de energía en modo de ionización de impacto electrónico (EMIE), debe ser capaz de producir un espectro de masas que cumpla con todos los criterios de la Tabla B.3-1, de este Apéndice, con 4-bromofluorobenceno (BFB).
Espátula de acero inoxidable Envases de 20 mL con tapón de rosca recubierto con PTFE Interface del CG/EM. Utilizar una interface de acoplamiento directo de la columna dentro del MS, las columnas generalmente utilizadas tienen un diámetro interno de 0.25 a 0.32 mm. Para columnas de 0.53 mm, utilizar un separador de jet, que incluye una línea de transferencia de todo el vidrio y dispositivo de vidrio de enriquecimiento o interface de división (Split). Esta interface es necesaria cuando se utiliza enfriamiento criogénico, la cual condensa los componentes desorbidos, sobre una banda estrecha de la pre-columna de sílice fundida no recubierta. Cuando la interface se calienta rápidamente, la muestra es transferida a la columna analítica capilar. Durante la etapa de crioconservación, la temperatura de la sílice fundida en la interface se mantiene a -150 ºC bajo una corriente de nitrógeno líquido. Después del periodo de desorción, la interface debe ser capaz de calentarse rápidamente (en 15 s o menos) a 250° C para completar la transferencia de los analitos. Jeringas de 0.5, 1.0, 5, 10 y 25 mL de vidrio hipodérmico con punta Luer. Jeringa con válvula de dos vías, con la terminación Luer, aplicable al dispositivo de purga Materiales de empaque para la trampa. Se recomienda el empaque de metil silicona pero puede utilizarse otro tipo de empaque que tenga la capacidad de retener los compuestos de interés (ver recomendaciones del fabricante. Matraces volumétricos de vidrio con tapón esmerilado de 10, 50 y 100 mL verificados previamente. Microjeringas de 10, 25, 100, 250, 500 y 1 000 µL Sistema de datos. Conectar al espectrómetro de masas una computadora que permita la adquisición y almacenamiento de todas las masas del espectro obtenido a través del programa cromatográfico. El software debe permitir la búsqueda de todos los espectros adquiridos para las masas (m/z) de los iones específicos y graficar las abundancias de tales m/z en función del tiempo o número de barridos. Este tipo de gráfico es definido como el PCIE. El software también debe permitir la integración de las abundancias de cualquier PCIE sobre un tiempo específico o un límite de scan. Siendo necesaria la versión más reciente de biblioteca de espectros. Sistema de purga y trampa o sistema de purga y trampa/headspace. El sistema de purga y trampa consiste en un dispositivo de purga, trampa y un desorbedor. Existen diferentes sistemas disponibles comercialmente. Trampa. De al menos 25 cm de largo y con un diámetro interno de al menos 3 mm, empacado con las siguientes longitudes mínimas de adsorbentes: 1.0 cm de empaque de metil silicona, 7.7 cm del polímero 2,6-difenilo óxido, 7.7 cm de sílica gel y 7.7 cm de carbón de coco, o algún otra propuesta del proveedor que nos brinde la concentración de los volátiles esperados y cumplan con todos los criterios de calidad. Existen trampas de sorbentes disponibles comercialmente. Las especificaciones del ejemplo para la trampa se ilustran en la Fig. B.3-2. 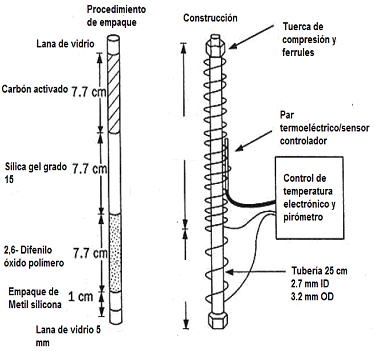 Fig. B.3-2. Especificaciones del ejemplo de trampa Válvulas de jeringa, bilateral, con punta desechable. Vaso de precipitados de 100 mL de vidrio. Viales de 40 mL con tapón de rosca recubierto con PTFE o Teflon B.3.5 Reactivos y soluciones Ácido clorhídrico, HCl. Grado reactivo que cumpla las especificaciones de la American Chemical Society (ACS) o equivalente. Libre de compuestos orgánicos volátiles y bajo condiciones de aseguramiento de los volátiles presentes en las muestras. Ácido clorhídrico 1:1 v/v. Adicionar cuidadosamente un volumen medido de HCl a un volumen igual de agua libre de compuestos orgánicos. Agua Tipo I. Agua libre de compuestos orgánicos. Carbón de coco Pasado a través de malla 26 (Material del empaque de la trampa). Clorobenceno d5 (Estándar interno). Empaque de metil-silicona-OV-1 (3%) En chromosorb-W, con malla 60/80 o equivalente (Material del empaque de la trampa). Estándares de Control de Calidad Gas inerte de alta pureza Materiales de referencia. Certificado (trazables) para benceno, tolueno, etilbenceno, Xilenos (orto, meta y para) y estireno. Metanol CH3OH. Libre de compuestos orgánicos Nitrógeno líquido (en caso necesario) Polímero 2,6-difenilo óxido. De malla 60/80, grado cromatográfico (Material del empaque de la trampa). Sílica gel. De malla 35/60, grado 15 o equivalente (Material del empaque de la trampa). Tolueno d8 (Estándar surrogado) 1,4 difluorobenceno d4 (Estándar interno) 4-bromofluorobenceno (Estándar surrogado) 1,2-dichloroethane-d4 (Estándar surrogado). Otros compuestos pueden ser usados como surrogados, mientras se demuestre su desempeño. B.3.6 Procedimiento B.3.6.1 Preparación de diluciones estándar B.3.6.1.1 Para preparar las disoluciones estándar concentradas de benceno, tolueno, etilbenceno, xilenos (orto, meta y para) y estireno, se debe preparar a partir de materiales estándar puros o de disoluciones certificadas. Preparar las disoluciones estándar en metanol. Colocar aproximadamente 9.8 mL de metanol en un matraz volumétrico de 10 mL que ha sido tarado incluyendo el tapón. Dejar reposar el matraz sin tapar, durante unos 10 minutos o hasta que todos las superficies humedecidas con alcohol se hayan secado. Pesar el matraz con una precisión de 0.1 mg. Adicionar los materiales de referencia usando una jeringa de 100 µL o una pipeta de vidrio con punta capilar desechable, inmediatamente adicionar dos o más gotas del material de referencia en el matraz. Asegurarse de que las gotas caigan directamente en el alcohol sin tocar el cuello del matraz. Después volver a pesar el matraz, llevar a volumen, tapar y agitar invirtiéndolo varias veces. Calcular la concentración en µg/L de ganancia neta en el pesaje. Cuando la pureza de un compuesto es 96%, calcular la concentración de la disolución sin considerar la pureza para el cálculo. Preferentemente usar estándares preparados comercialmente certificados por el fabricante o una fuente independiente. Transferir la disolución estándar a un envase con tapón de rosca recubierto con PTFE. Almacenar con un mínimo espacio de cabeza (headspace) y protegido de la luz a una temperatura 6 °C. B.3.6.1.2 Para preparar las disoluciones estándares de trabajo de benceno, tolueno, etilbenceno, xilenos (orto, meta y para) y estireno, a partir de las disoluciones de estándares concentradas, preparar disoluciones multicomponentes en metanol a una concentración adecuada, almacenar con un mínimo de espacio de cabeza, y revisada frecuentemente por signos de degradación o evaporación. Siendo necesario reemplazarlos después de 2 a 4 semanas a menos que se tenga documentada el funcionamiento adecuado del mismo. Si se trabaja con soluciones de mezclas certificadas, se pueden almacenar de acuerdo a las indicaciones del proveedor. Para la preparación de está disolución metanólica no utilizar un volumen menor de 10 µL. B.3.6.1.3 Para preparar las disoluciones estándares de trabajo del surrogado/ estándar interno, los compuestos surrogados recomendados son: tolueno d8, 4-bromofluorobenceno y 1,2- dicloroetano-d4, pueden utilizarse otros compuestos dependiendo de los requerimientos del análisis y de los analitos que se requieren cuantificar. Es necesario preparar una solución madre y de ella la solución apropiada para adicionar. Cada muestra que se va analizar debe contener los subrogados. Los estándares internos recomendados son el fluorobenceno, clorobenceno-d5 y el 1,4-diclorobenceno-d4, se pueden utilizar otros compuestos siempre y cuando tengan tiempos de retención similares a los analitos detectados. Es necesario preparar una solución madre y de ella la solución apropiada para adicionar. Se recomienda una solución de 25 mg/L de cada estándar interno, de modo que la adición de 10 µL a una muestra de 5 mL sería equivalente a 50 µg/L. Si se emplea un espectrómetro de masas más sensible se puede emplear una solución de estándares internos más diluida. Las áreas de los estándares internos deberán están entre 50 y 200% de las áreas de los analitos en los puntos medios de las curvas de calibración. B.3.6.1.4 Para preparar el estándar de BFB, preparar una solución de estándar conteniendo 25 ng/µL de BFB en metanol, si se emplea un espectrómetro de masas más sensible, se puede emplear una solución más diluida. B.3.6.1.5 Para las disoluciones acuosas de los estándares de calibración, se debe de construir una curva de calibración para cada analito (BTEX y estireno) de cinco a siete concentraciones. La disolución estándar de menor concentración deberá ser mayor al límite de detección pero muy cercano. Preparar cada una de las mezclas de disoluciones estándar de la curva de calibración inyectando rápidamente el volumen requerido de la disolución estándar metanólica (mezcla de estándares de BTEX y estireno, en el matraz volumétrico previamente lleno con agua libre de compuestos orgánicos, sumergiendo la aguja dentro del agua, se recomienda inclinar el matraz mientras se inyecta la mezcla de estándares. Posteriormente adicionar a cada matraz las soluciones de estándar interno y estándar surrogado. B.3.6.2 Procedimiento analítico Para la optimización del CG/EM, las condiciones cromatográficas dependen de los compuestos de interés, el instrumento y los proveedores de las columnas. Las condiciones cromatográficas recomendadas para inyección directa (ejemplo) son: · Temperatura de inyector: 200-275 °C · Temperatura de la línea de transferencia: 200-300 °C · Columna cromatográfica: DB-624 (6%cianopropilfenil/94%dimetipolisiloxano) de 70 m X 0.53 mm o cualquier columna equivalente que proporcione un desempeño de método que permita cumplir con las especificaciones de esta Norma. · Flujo de gas acarreador 4 ml/min · Programación de condiciones: temperatura inicial 40 °C durante 3 minutos, 8 °C/minuto hasta 260 °C, hasta que todos los compuestos esperados hayan eluído. · Bake column durante 75 minutos. De manera general las condiciones de análisis estarán en función de los equipos e instrumento utilizado para la determinación (éstos incluye cromatógrafo, detector, columna cromatográfica, etc.). El espectrómetro de masas requiere un ajuste adicional, sintonizando de tal manera que al comienzo de cada periodo de 12 horas de análisis y antes del análisis de blancos, estándares y muestras verificar el sistema CG/EM inyectando 25 ng de 4-bromofluorobenceno (BFB) directamente en la columna del cromatógrafo. Si se dificulta la inyección directa, adicionar 1 µL de disolución de BFB de 25 µg/mL a 25 mL de agua en una jeringa utilizada para transferir muestras para dispositivos de purga y analizar como una muestra. Se debe de obtener un espectro de masas de corrección de fondo y confirmar que se cumplen los criterios de abundancia m/z descritos en la Tabla B.3-1. Si no se cumplen todos los criterios, repetir la prueba hasta que se cumplan todos. En relación con el procedimiento de purga y trampa, es necesario llevar a cabo el análisis para la purga y concentración de los analitos a temperatura ambiente. Es necesario que los estándares de calibración, muestras y muestras control de calidad sean purgadas a la misma temperatura. Purgar la muestra con helio u otro gas inerte a un flujo entre 20 y 40 mL/minuto durante 11 minutos. Llevar a cabo la desorción utilizando una temperatura de 245 °C con un flujo de 10 mL/min y una duración de 1.5 minutos o de acuerdo a las condiciones establecidas durante la optimización del método. Después de la desorción de la muestra reacondicionar la trampa reiniciando el ciclo de purga y manteniendo la temperatura de la trampa a 245 °C durante 10 minutos. Proceder de igual manera con los blancos, curva de calibración y muestras de control de calidad. De manera general las condiciones de análisis estarán en función del equipo utilizado para la determinación (sistema de purga y trampa y trampa utilizada). B.3.6.3 Análisis de la muestra Las muestras deben almacenarse en viales con tapa con un espacio mínimo de cabeza, a 4 °C o menos y en un área libre de disolventes orgánicos. El tamaño de alguna burbuja formada durante el enfriamiento de la muestra no debe ser mayor de 5-6 mm, cuando se observe alguna burbuja en el vial se debe revisar que éste no presente fuga, de ser así la muestra debe descartarse. Todas las muestras deben analizarse tan pronto como sea posible, después de haber sido recolectadas, generalmente en un máximo de 14 días después de la recolección. Poner a temperatura ambiente la muestra y transferir 5mL al dispositivo de purga utilizando el automuestreador, o manualmente utilizando una jeringa. Si los volúmenes de muestra son mayores de 5 mL, por ejemplo muestras de 25 mL, entonces los volúmenes de los estándares de calibración deben ser los mismos. Para tomar la alícuota de la muestra que será inyectada en el sistema de purga en forma manual, utilizar una jeringa con válvula de cierre. Retirar el émbolo de la jeringa y cerrar la válvula. Abrir el envase de la muestra y cuidadosamente colocar la muestra dentro del cuerpo de la jeringa. Colocar nuevamente el émbolo de la jeringa y antes de presionar el émbolo, invertir la jeringa y abrir la válvula, posteriormente presionar el émbolo para eliminar el aire y ajustar el volumen de la muestra a 5 ó 25 mL según sea el caso. Si hay muestra suficiente disponible, utilizar otra jeringa para tomar una segunda alícuota, para su análisis por duplicado (una vez que la tapa de la muestra se ha removido, la muestra no se puede almacenar, debido al espacio de cabeza (headspace). Adicionar una cantidad apropiada de la mezcla de los estándares surrogados y estándar interno a través del orificio de la válvula y cerrarla. Unir con el dispositivo de purga, abrir las válvulas e inyectar la muestra en el recipiente de purga. Cerrar las válvulas y purgar la muestra por 11 minutos a temperatura ambiente a un flujo de 40 mL/min (helio o nitrógeno). Si el vapor de agua causa problemas en el espectrómetro de masas, utilizar una purga seca de 3 minutos y/o un módulo de control de humedad. Absorber los materiales atrapados en la cabeza de la columna cromatográfica a 180 °C mientras pasa el gas inerte a un flujo compatible con la columna de elección y comenzar un programa de temperatura en el CG. Establecer el sistema de auto-drenaje para la cámara de purga vacío mientras la trampa está siendo absorbida dentro del CG, o alternativamente, utilizar una jeringa de muestra para vaciar el recipiente. Lavar la cámara con dos enjuagues de 25 mL de agua si las muestras que se están analizando están altamente contaminadas. Asegurarse que todas las áreas humedecidas durante la purga están también humedecidas durante el enjuague para maximizar el enjuague. Reacondicionar la trampa en el horno a la temperatura de acuerdo a lo que recomienda el fabricante por 5 a 7 minutos. Dejar que la trampa se enfríe a temperatura ambiente antes de introducir la siguiente muestra en el recipiente de purga. Cuando todos los compuestos de la muestra han sido eluidos de la columna cromatográfica, terminar la adquisición de datos y guardar los archivos. Usar un software que muestre el intervalo completo de los espectros de masas y un apropiado EICP. Si la abundancia de un ion excede el sistema del intervalo de trabajo, diluir la muestra en una segunda jeringa con agua y analizar. Tener cuidado con la muestra porque los compuestos pueden ser volátiles y se pueden perder si se reabre la muestra. Estimar la dilución necesaria y expulsar el exceso de muestra de la segunda jeringa, inyectar esa porción en un recipiente de purga y con una segunda jeringa, adicionar el agua necesaria para tener un total de 25 mL en el recipiente de purga. B.3.6.4 Análisis de datos y cálculos Una vez que los compuestos han sido identificados, la cuantificación de dichos compuestos se basa en la abundancia del área integrada del PCIE, el estándar interno utilizado debería tener el tiempo de retención muy cercano al analito de interés. B.3.6.5 Informe de prueba Reportar cada uno de los compuestos (benceno, tolueno, o-xileno, meta-xileno, para-xileno y estireno) en µg/L. B.3.7 Control de calidad, calibración e interferencias B.3.7.1 Control de calidad Antes del análisis de las muestras leer un blanco para demostrar que todas las partes del equipo en contacto con la muestra y los reactivos están libres de interferencias. Los blancos de reactivos, blancos de almacenamiento, muestras fortificadas y duplicados deben someterse a los mismos procedimientos analíticos que los utilizados en las muestras. El laboratorio debe demostrar la competencia inicial y mantener los registros de esto. Durante el análisis de rutina, se deben analizar blancos de reactivos, blancos de almacenamiento, muestras fortificadas y duplicados de muestras. Se deben agregar surrogados a las muestras y a la muestras de control de calidad. Siempre que sea posible, el laboratorio debe analizar materiales de referencia certificados y participar en estudios de pruebas de aptitud. El laboratorio debe tener procedimientos de control de calidad para asegurarse de que la integridad de la muestra no se vea comprometida durante el proceso de recolección y manipulación de las muestras, por ejemplo, asegurándose que las tapas y septas de los viales no presenten fugas. El espectrómetro de masas se calibra con el estándar de BFB ajustando masas e intensidad de los iones. Los tiempos de retención relativos de los analitos en la muestra deben estar dentro de ± 0.06 unidades de tiempo de retención de los estándares. La intensidad relativa de los iones característicos de cada uno de los compuestos en la muestra debe encontrarse dentro del 30% de las intensidades relativas de estos iones en el espectro de los estándares de referencia (ejemplo: para un ion con abundancia del 50% en el espectro de referencia, la abundancia en el espectro de la muestra debe encontrarse en el intervalo de 20% al 80%). La curva de calibración inicial para cada compuesto de interés debe verificarse cada 12 horas, analizando un estándar de calibración de concentración cercana al punto medio del intervalo de la curva de calibración (CdC). Si el %DSR de los factores de respuesta es menor o igual al 20%, la calibración inicial continúa vigente, si el valor es mayor que 20% para cualquier CdC, revisar la preparación del estándar, las condiciones instrumentales, si el problema continúa preparar una nueva curva y recalibrar. Analizar un blanco de reactivos después de analizar el estándar de calibración o intermedio en el lote analítico, para asegurar que el sistema está libre de contaminación. B.3.7.2 Calibración B.3.7.2.1 Preparar una curva de calibración de al menos cinco niveles de concentración conteniendo las concentraciones requeridas de estándares (BTEX y estireno) de calibración, incluyendo subrogados y/o estándares internos, y transferir 5 ó 25 mL (según se requiera) al dispositivo de purga utilizando el automuestreador, o manualmente utilizando una jeringa. Si los volúmenes de muestra son mayores de 5 mL, por ejemplo muestras de 25 mL, entonces los volúmenes de los estándares de calibración deben ser los mismos. El cálculo de regresión debe generar un coeficiente de correlación (r) mayor o igual a 0.99. B.3.7.2.2 Procedimiento de calibración con estándar externo Emplear este procedimiento solo si el volumen de la inyección puede mantenerse constante. Analizar cada concentración de la curva de calibración. Calcular individualmente los factores de respuesta (FR) para cada estándar analizado de la siguiente manera: 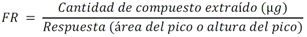 Calcular la cantidad de compuesto para cada estándar: 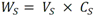 En donde: WS cantidad del compuesto, µg VS volumen del estándar extraído, L CS concentración del estándar preparado en µg/L Para cada componente determinar el promedio del factor de respuesta (FR) y la desviación estándar de los factores de respuesta empleando todos los estándares de calibración analizados. Si el porciento de la desviación estándar relativa (%DER) es menos del 20%, se puede emplear el FR promedio para calcular la concentración de la muestra: 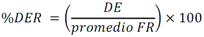 En donde: DE desviación estándar FR factor de respuesta Si él %DER es mayor del 20 %, graficar la curva de calibración de cantidad inyectada contra respuesta, emplear la gráfica para determinar la cantidad del componente presente en cada muestra. Después determinar la concentración dividiendo la cantidad, µg, por el volumen en L, de muestra extraída. Opcionalmente emplear el sistema de datos para realizar una regresión lineal y emplear la ecuación para calcular la cantidad de los componentes a partir de los valores de respuesta. Cuando se emplee el valor promedio del FR, calcular la concentración como sigue: 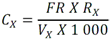 En donde: CX concentración del compuesto en mg/L RX respuesta de la muestra (mm, área, etc.) VX volumen de la muestra extraída en L B.3.7.2.3 Procedimiento de calibración con estándar interno. Emplear este procedimiento cuando el volumen de la inyección no se mantiene constante. Los estándares internos deben ser los más cercanos a los analitos a cuantificar de modo que tengan tiempos de retención relativos de 0.80 1.20. Usar el ion del pico base de estándar interno específico como ion primario para la cuantificación. Si se presentan interferencias utilice el siguiente ion de mayor intensidad para la cuantificación. Elaborar una lista con las áreas de los iones característicos (Tabla B.3-1, de este Apéndice), contra la concentración de cada compuesto y estándar interno. Calcular los FRR para cada compuesto relativo a cada uno de los estándares internos.
Analizar cada concentración de la curva de calibración. Para todos los análisis hechos en una secuencia analítica, determinar el promedio y la desviación estándar de la respuesta del estándar interno. Calcular el porciento de la desviación estándar relativa (%DER), si el %DER es mayor del 20%, tomar acciones correctivas para mejorar la precisión del método. Establecer un intervalo de confianza de 99% para la respuesta del estándar interno usando el promedio calculado y la desviación estándar del análisis de la muestras. Rechazar los análisis cuando la respuesta del estándar interno está fuera de los límites y volver a analizar. Después del análisis de los estándares de calibración, calcular los factores de respuesta relativos (FRR) individuales de cada analito en cada estándar de la siguiente manera: 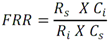 En donde: Rs, Ri respuestas del estándar de calibración y del estándar interno respectivamente. Cs, Ci concentraciones del analito en los estándares de calibración y del estándar interno respectivamente. Calcular el promedio de los FRR de cada analito, las desviaciones estándar de los FRR y %DER. Si él %DER es menor del 20%, utilizar el valor promedio del FRR; si es mayor, graficar la curva de calibración o la ecuación de regresión lineal como se describe en el procedimiento de estándar externo. Cuando se utilice el promedio de los FRR del estándar interno, calcular la concentración de las muestras con la ecuación siguiente: 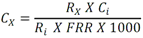 En donde: Cx concentración del analito en la muestra en mg/L Rx respuesta de la muestra Ci concentración del estándar interno Ri respuesta del estándar interno B.3.7.3 Interferencias Las interferencias introducidas por las muestras son contaminantes que son co-extraídos de ellas por lo que la cantidad de estas varía dependiendo del tipo de muestra y su naturaleza. Las interferencias del método pueden originarse por la presencia de contaminantes en el material de vidrio o en el sistema de purga y trampa que puede conducir a obtener picos distorsionados y picos fantasma y/o línea base elevada en el cromatograma. El blanco electrónico (inyección de aire) permite verificar el grado de contaminación del sistema. Tomar en consideración las impurezas en el gas de purga y compuestos orgánicos de desgasificación. También puede ser ocasionada por la contaminación del equipo y de la tubería de la trampa. Demostrar que el sistema está libre de contaminación bajo condiciones de operación analizando los blancos de reactivos diariamente. Utilizar blancos sólo para monitorear; son inaceptables las correcciones por valores de blancos. En el sistema de purga y trampa evitar usar tubería de plástico y selladores de rosca que no sean de PTFE, o controladores de flujo con componentes de caucho. Asegurarse que el área analítica no es fuente de contaminación por disolventes de laboratorio. Utilizar un blanco de reactivos preparado a partir de agua y llevado a través de la toma de muestras, manipulación y procedimientos de envío como un control de este tipo de contaminación. La contaminación por arrastre puede ocurrir siempre que se analiza una muestra con concentraciones altas del o los analitos e inmediatamente después se analiza una muestra con concentraciones bajas. Para reducir el arrastre enjuagar el dispositivo de purga y la jeringa de inyección de muestra con agua entre muestra y muestra. Después del análisis de una muestra de concentración alta, analizar un blanco de reactivos para verificar que esté libre de contaminación. Para muestras que contienen cantidades de materiales solubles en agua, sólidos suspendidos, compuestos con alto punto de ebullición o altos niveles de compuestos volátiles, entre análisis lavar la cámara de purga con una solución detergente, enjuagar con agua destilada y secar en un horno a 105 °C. La trampa y otras partes del sistema también son objeto de contaminación, por lo tanto frecuentemente purgar todo el sistema. B.3.8 Seguridad La toxicidad o carcinogenicidad de todos los reactivos no se han determinado con precisión. Sin embargo, todas las sustancias deben ser tratadas como potencial peligroso para la salud, por lo que al manipular las disoluciones estándar, los analistas deberán realizarlo en campana y utilizar respiradores con filtros especiales para gases tóxicos además del vestuario básico (bata y guantes). Estos materiales estándar puros y las disoluciones preparadas deberán almacenarse en condiciones recomendadas por el proveedor. B.3.9 Referencias EPA Method 8260c Volatile Organic Compounds by gas Chromatography/mass Spectrometry (GC/MS) EPA Method 5030c Purge and Trap for Aqueous Samples. B.4 MÉTODO DE PRUEBA PARA LA DETERMINACIÓN DE PLATA TOTAL Y ALUMINIO EN AGUA PARA USO Y CONSUMO HUMANO Para la determinación de plata total y aluminio para el cumplimiento de esta Norma, se podrá utilizar indistintamente el método de prueba para la determinación de plata total y aluminio en agua para uso y consumo humano por espectrometría de absorción atómica con horno de grafito (punto B.4.1, de este Apéndice) o el método de prueba para la determinación de aluminio, antimonio, arsénico, bario, cadmio, cobre, cromo, fierro, manganeso, níquel, plata, plomo y selenio por espectrometría de emisión óptica de plasma acoplado inductivamente (ICP-OES). (punto B.4.2, de este Apéndice). B.4.1 MÉTODO DE PRUEBA PARA LA DETERMINACIÓN DE PLATA TOTAL Y ALUMINIO EN AGUA PARA USO Y CONSUMO HUMANO POR ESPECTROMETRÍA DE ABSORCIÓN ATÓMICA CON HORNO DE GRAFITO B.4.1.1 Símbolos y términos abreviados Ag plata Al aluminio HCl ácido clorhídrico LCH lámpara de cátodo hueco LDE lámpara de descarga sin electrodos PTFE politetrafluoroetileno B.4.1.2 Principio Las muestras de agua se conservan mediante tratamiento con ácido y se digieren si es necesario. Una pequeña submuestra se inyecta en el horno de grafito del espectrómetro de absorción atómica. El horno se calienta eléctricamente. Al aumentar la temperatura gradualmente, la muestra se seca, se piroliza y se atomiza. La espectrometría de absorción atómica se basa en la capacidad de los átomos para absorber la luz de una fuente de luz que emite una luz específica para un cierto elemento. Cuando el haz de luz pasa a través de la nube de átomos en el horno de grafito calentado, la luz es absorbida selectivamente por los átomos de los elementos elegidos. La disminución de la intensidad de la luz se mide con un detector a una longitud de onda específica. La concentración de un elemento en una muestra se determina comparando la absorbancia de la muestra con la absorbancia de disoluciones de calibración. B.4.1.3 Alcance y aplicación El método descrito es un procedimiento para la determinación de niveles trazas de plata total (Ag) y aluminio (Al) en agua de uso y consumo humano proveniente de sistemas de abastecimiento utilizando la espectrometría de absorción atómica con atomización electrótérmica en horno de grafito. El límite de detección del método para plata es de 0.2 µg/L. El límite de detección del método para aluminio es de 1 µg/L. Este método es aplicable también para la determinación de bajas concentraciones de Arsénico (As), Cadmio (Cd), Cobalto (Co), Cromo (Cr), Cobre (Cu), Fierro (Fe), Manganeso (Mn), Molibdeno (Mo), Níquel (Ni), Plomo (Pb), Antimonio (Sb), Selenio (Se), Talio (Tl), Vanadio (V) y Zinc, para agua de uso y consumo humano proveniente de sistemas de abastecimiento, así como para agua superficial y subterránea, sin embargo, este documento se refiere exclusivamente para la determinación de plata total y aluminio en agua de uso y consumo humano proveniente de sistemas de abastecimiento, por lo que para la determinación de otros elementos y en otras matrices deberán de realizarse las adecuaciones necesarias. B.4.1.4 Equipos y materiales Las referencias a marcas específicas y números de catálogo se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden representar especificaciones adecuadas para los artículos. El límite de detección depende de la matriz de la muestra así como del equipo, el tipo de atomizador y el uso de modificadores químicos. Para muestras de agua con una matriz simple (concentración baja de sólidos y partículas disueltas), los límites de detección del método estarán cerca de los límites de detección del equipo. Balanza analítica. Con sensibilidad de 0.1mg Centrífuga de laboratorio. Capaz de mantener 1600 rpm Embudos de filtración. De diferentes capacidades: de polietileno, polipropileno, PTFE ó vidrio. Espectrómetro de absorción atómica con horno de grafito. Equipado con sistema de corrección de fondo y automuestreador. Filtros. Con tamaño de poro de 0.45 µm Fuente de radiofrecuencia. En caso de usar LDE. Horno de microondas. O sistema de reflujo o sistema de digestión abierto. Lámpara. De cátodo hueco (LCH) y/o Lámpara multi-elemento y/o lámpara de descarga sin electrodos (LDE) de plata. Material común de laboratorio. Matraces volumétricos. De diferentes capacidades. Papel filtro. De filtración media # 1, 2, 111, 43, 41, 40 etc. Pipetas de pistón (micropipetas). De diferentes capacidades. Puntas de plástico. Para pipetas de pistón. Recipientes. De polipropileno o PTFE. Sistema de extracción. Para eliminar el humo y los vapores que son peligrosos para la salud del analista. Tubos de grafito. Cubiertos pirolíticamente Viales B.4.1.5 Reactivos y soluciones Ácido nítrico (HNO3). Grado reactivo (G.R) Ácido nítrico de alta pureza (HNO3). Al 65% v/v (con contenido de metales en niveles traza). Agua tipo I. Argón o nitrógeno. De alta pureza grado absorción atómica. Disolución de ácido nítrico. Grado reactivo al 10% (Para la descontaminación del material). Por cada litro a preparar, agregar 100 mL de ácido de la siguiente manera: en un recipiente que contenga aproximadamente 2/3 partes de agua agregar la cantidad de ácido requerida y llevar al volumen total con agua. Disolución de nitrato de Magnesio hexahidratado [Mg (NO3)26H2O)]. De alta pureza (con contenido de metales a niveles traza). Disolución estándar de plata o aluminio. De 1000 mg/L de preferencia trazable a patrones nacionales o internacionales. La disolución puede prepararse a partir de la sal, y debe tener una pureza >99.95%. La sal debe ser secada por 1 h a 105 °C. Disolución de paladio (Pd). Con niveles traza de metales (de alta pureza). Fosfato de amonio monobásico (NH4H2PO4). Con niveles traza de metales (de alta pureza). B.4.1.6 Procedimiento B.4.1.6.1 Lavado de material Todo el material utilizado debe someterse a lavado de acuerdo con las siguientes instrucciones: El jabón que se use debe ser neutro de preferencia líquido. Enjuagar perfectamente con agua corriente. Sumergir el material de vidrio o plástico en un recipiente (de preferencia de plástico) que contenga una solución de ácido nítrico al 10%. El HCl es más efectivo para el lavado de material de polietileno o polipropileno, mientras que el HNO3 es preferible para el material de PTFE o de vidrio. Dejarlo tapado y reposando por un lapso mínimo de 2 horas. Quitar el exceso de ácido nítrico con varios enjuagues de agua de red y el último enjuague con agua tipo I. Dejar escurrir y secar. Tapar con papel parafinado o en bolsas cerradas de plástico. Guardar en cuanto esté seco para evitar contaminación por partículas en el aire. B.4.1.6.2 Preparación de las muestras Las muestras se pueden analizar directamente por espectrometría de absorción atómica sin realizar la digestión si son inodoras, incoloras y transparentes. Previo al análisis adicionar a 100 mL de muestra, 1 mL de ácido nítrico de ultra alta pureza. En caso que se observe un precipitado, este volumen de muestra se digiere adicionando 1 mL más de ácido nítrico de ultra alta pureza concentrado, calentar a 85°C hasta reducir el volumen a 20 mL cuidando que no hierva. En caso de que el precipitado sea considerable se sugiere la adición de 10 mL de ácido nítrico de ultra alta pureza para realizar la digestión de la muestra. Calentar a reflujo por 20 min y transferir a un matraz volumétrico de 50 mL. Llevar al volumen con agua tipo I Centrifugar a 1600 rpm por 30 minutos o dejar reposar toda la noche y analizar el sobrenadante. Se puede utilizar el horno de microondas para digerir las muestras si se forma un precipitado al adicionar el ácido nítrico. Proceder de acuerdo a las condiciones recomendadas por el fabricante. Preparar un blanco de reactivos fortificado por cada lote, y una muestra fortificada por cada 10 muestras o por grupo si son menos. B.4.1.6.3 Preparación de diluciones y curvas de calibración Para la preparación de las disoluciones madre (más concentradas). Medir un volumen apropiado de disolución estándar (aprox. 1 a 10 mL) para el aforo inicial, y hacer las diluciones necesarias utilizando material volumétrico verificado para su preparación. Para la preparación de las disoluciones más concentradas utilizar HNO3 de alta pureza de tal forma que la concentración final del ácido sea del 2 al 5% para poder preservar las disoluciones estándar por mayor tiempo, mantener estas, bien tapadas y en recipientes de PTFE de preferencia. Para la preparación de las disoluciones de trabajo y de la curva de calibración utilizar HNO3 de alta pureza y la concentración final del ácido debe estar entre 0.1 a 0.2%. Preparar éstas el mismo día del análisis. Para preparar la solución de trabajo de plata (Ag), disolver la plata en 80 ml de HNO3 (1+1) calentando para lograr la disolución, enfriar y diluir al volumen con agua en un matraz volumétrico de 1000mL. Reservar la solución en una botella cambar sellada completamente con una hoja de aluminio para proteger de la luz. Para preparar la solución de trabajo de aluminio (Al), disolver el aluminio en 4 mL de HCl (1+1) y 1 mL de HNO3 concentrado en un cubo. Calentar el cubo lentamente para inducir la solución. Transferir la solución cuantitativamente a un matraz volumétrico de 1000 mL. Añadir 10mL de HCl (1+1) y diluir al volumen deseado con agua. Preparar 5 niveles de concentración dentro del intervalo de trabajo. El intervalo depende de la sensibilidad del instrumento, del tipo de matriz y del uso de modificadores, tomar como guía la Tabla B.4.1-1, de este Apéndice, para un volumen de muestra de 20 µL.
Preparar una disolución de una concentración conocida de plata o aluminio para verificar el equipo. B.4.1.6.4 Condiciones de operación del horno de grafito Ajustar el espectrofotómetro de acuerdo a las recomendaciones del fabricante, encender la lámpara y dejar calentar al menos 15 minutos las LCH y al menos 45 minutos las LDE. Siempre utilizar la corrección de fondo. El programa de temperatura del horno de grafito (secado, pirolisis, atomización y limpieza) depende del analito, de la matriz y de la marca del equipo utilizado. Optimizar utilizando como guía las recomendaciones del fabricante. En la Tabla B.4.1-2, de este Apéndice, se muestran los principales parámetros utilizados para ajustar el equipo.
Utilizar modificadores de matriz para eliminar los efectos de matriz ya que su uso permite elevar la temperatura de pirolisis y poder eliminar las interferencias sin que se pierda el analito. Si se utilizan modificadores de matriz en las muestras, adicionar también al blanco de la curva, a la curva de calibración, a las disoluciones estándar de verificación, a las muestras fortificadas y a las disoluciones estándar de control de calidad (muestras de control de calidad (MCC) o muestras de control de laboratorio (MCL). En la Tabla B.4.1-3, de este Apéndice, se muestran los principales modificadores de matriz utilizados en el análisis por horno de grafito. Generalmente la cantidad indicada en la tabla debe estar contenida en un volumen de modificador de 10 µL.
Si el equipo cuenta con automuestreador, colocar los puntos de la curva, el blanco de reactivos, las muestras y los modificadores de matriz en los viales, los cuales han sido previamente enjuagados con ácido nítrico al 3% y posteriormente con la disolución a analizar. B.4.1.6.5 Lectura en el equipo Leer por triplicado el blanco de calibración para verificar que no haya contaminación y posteriormente leer la disolución de verificación también por triplicado. Leer por duplicado en el equipo el blanco y los puntos de la curva. Elaborar una curva de calibración dentro del intervalo de trabajo, graficando la absorbancia (área o altura de pico) en función de la concentración. La absorbancia integrada como área de pico es más recomendable. Ajustar la curva por medio de mínimos cuadrados (regresión lineal) o calcular la concentración directamente en el equipo que se programe. Leer cada una de las muestras por duplicado, registrar la absorbancia y calcular la concentración del elemento a partir de la curva de calibración. Cuando se use el equipo programable realizar los cálculos finales. Asegurarse que las concentraciones de las muestras caen dentro del intervalo de trabajo de la curva de calibración, de no ser así realizar la dilución correspondiente. B.4.1.6.6 Análisis de datos y cálculos Interpolar los valores de absorbancia, área o altura del pico de la muestra analizada en la curva de calibración y obtener los resultados en mg/L del elemento en la muestra empleando la siguiente fórmula:  En donde: A concentración del elemento en la muestra leída directamente del equipo o de la curva de calibración, en mg/L o µg/L V volumen de la disolución de la muestra (aforo), en mL F factor para pasar de µg a mg=1 000 (si la curva está en µg/L) M volumen de la muestra, en mL Si la muestra ha sido diluida, debe aplicarse el factor de dilución. Calcular el porcentaje de recuperación para el analito de acuerdo con la siguiente fórmula: 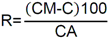 En donde: R % de recobro CM Concentración de la muestra fortificada o blanco de reactivos fortificado. C Concentración de la muestra o blanco de reactivos. CA Concentración equivalente de elemento añadido a la muestra o blanco de reactivos. B.4.1.6.7 Informe de prueba Reportar el resultado como mg/L de plata total o mg/L de aluminio. B.4.1.7 Control de calidad e interferencias B.4.1.7.1 Control de calidad El coeficiente de correlación (r) de la curva deberá ser > 0.995. Leer en el equipo el blanco de calibración y un punto de la curva de calibración (MCI) antes de iniciar la lectura de las muestras, después de la lectura de cada 10 muestras y al final del análisis. El resultado deberá encontrarse dentro del ± 15% del valor esperado. Si dicho valor no se encuentra en el intervalo, interrumpir el análisis y buscar las posibles causas, posteriormente volver a leer la curva de calibración y repetir las lecturas del último lote de muestras. B.4.1.7.2 Interferencias Altas concentraciones de cloruro pueden causar resultados bajos debido a que la volatilidad de muchos elementos se incrementa y el analito se pierde durante el proceso de pirolisis. Los efectos de matriz pueden disminuirse parcialmente o completamente con la optimización del programa de temperatura, del uso de tubos recubiertos pirolíticamente y plataformas, con el uso de modificadores químicos como el nitrato de magnesio o el fosfato de amonio, la técnica de adición de estándar y con el uso del corrector de fondo. B.4.1.8 Referencias International Organization for Standardization. 2003. Water quality- Determination of trace elements using atomic absorption spectrometry with graphite furnace. ISO15586:2003(E). B.4.2 MÉTODO DE PRUEBA PARA LA DETERMINACIÓN DE ALUMINIO, ANTIMONIO, ARSÉNICO, BARIO, CADMIO, COBRE, CROMO, FIERRO, MANGANESO, NÍQUEL, PLATA, PLOMO Y SELENIO POR ESPECTROMETRÍA DE EMISIÓN ÓPTICA DE PLASMA ACOPLADO INDUCTIVAMENTE (ICP-OES). B.4.2.1 Símbolos y términos abreviados Al aluminio Ag plata As arsénico Ba bario CCC verificación de calibración continua Cd cadmio Cr cromo CVI disolución estándar de verificación de la calibración inicial Fe fierro FEP fluorocarbono HCl ácido clorhídrico ICP-OES Espectrometría de emisión óptica de plasma acoplado inductivamente IEC interferencias espectrales LDI límites de detección del instrumental Mn manganeso Ni níquel Pb plomo Sb antimonio Se selenio Y itrio B.4.2.2 Principio La muestra en solución es aspirada (nebulizada) continuamente en un plasma de argón inductivamente acoplado, donde los analitos de interés se convierten en átomos de fase gaseosa de estado excitado o iones. A medida que los átomos o iones de estado excitado vuelven a su estado fundamental, emiten energía en forma de luz a longitudes de onda que son características de cada elemento. La intensidad de la energía emitida a la longitud de onda elegida es proporcional a la cantidad del elemento en la muestra analizada. Las muestras de agua se conservan mediante tratamiento con ácido y se digieren si es necesario. El análisis directo por ICP-OES debe llevarse a cabo solamente sobre matrices acuosas relativamente limpias. B.4.2.3 Alcance y aplicación El método de Espectrometría de emisión óptica de plasma acoplado inductivamente (ICP-OES) utilizando sistemas ópticos simultáneos o secuenciales y un sistema de plasma de vista axial y radial, es utilizado para la determinación de metales, metaloides y algunos elementos no metálicos en disolución acuosa. La espectrometría de emisión óptica de plasma acoplado inductivamente (ICP-OES) es una técnica espectrométrica utilizada para determinar oligoelementos en soluciones acuosas. En la ICP-OES, una solución de muestra es aspirada (es decir, nebulizada) continuamente en una descarga de argonplasma acoplada inductivamente, donde los analitos de interés se convierten en átomos o iones en fase gaseosa de estado excitado. A medida que los átomos o iones de estado excitado vuelven a su estado fundamental, emiten energía en forma de luz a longitudes de onda que son características de cada elemento específico. La intensidad de la energía emitida a la longitud de onda elegida es proporcional a la cantidad (concentración) de ese elemento en la muestra analizada. Por tanto, determinando las longitudes de onda que son emitidas por una muestra y sus intensidades respectivas, se puede cuantificar la composición elemental de la muestra dada con respecto a un patrón de referencia. Para obtener resultados precisos, el análisis directo de ICP-OES debe llevarse a cabo solamente sobre matrices acuosas relativamente limpias (por ejemplo, muestras de agua subterránea pre-filtradas). Otras muestras acuosas y/o sólidas más complejas necesitan digestión ácida antes de su análisis; El analista debe asegurar que se elija un método de digestión de la muestra que sea apropiado para cada analito y el uso previsto de los datos. B.4.2.4 Equipos y materiales Las referencias a marcas específicas y números de catálogo se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden representar especificaciones adecuadas para los artículos. Automuestreador. Balanza analítica, para la preparación de estándares y reactivos. Bomba peristáltica para introducir los estándares y las muestras al nebulizador. Control automático de los flujos de argón para control exacto y poder reproducir las condiciones del plasma. Embudos Espectrómetro de emisión controlado a través de una computadora, con capacidad de realizar corrección de fondo. Generador de radiofrecuencia Matraces volumétricos de diferentes capacidades. Nebulizador ultrasónico Pipetas volumétricas de pistón de diferentes capacidades. Espectrometría de emisión óptica de plasma acoplado inductivamente (ICP-OES). Probetas graduadas Unidad de microondas que proporcione una energía de 600 a 1600 W, dependiendo del número de muestras de capacidad, que pueda programarse dentro de ±10 W de la energía requerida y que cuente con sensor de temperatura y presión y un controlador de microondas. El sistema requiere de vasos de digestión de teflón PFA de 75 a 100 mL de capacidad capaces de resistir presiones de 500 psi como máximo y una temperatura máxima de 210 °C. B.4.2.5 Reactivos y soluciones A menos que se indique otro grado, los reactivos que se requieren en el método deben ser tipo ACS (American Chemical Society) grado reactivo analítico. Prever que los reactivos sean de la pureza necesaria para permitir su uso sin que afecte la exactitud en la determinación. Agua tipo I Ácido clorhídrico concentrado (HCl) con la pureza necesaria para evitar contaminación de las muestras. Ácido nítrico concentrado (HNO3) con la pureza necesaria para evitar contaminación de las muestras. Disolución de ácido clorhídrico HCl (1:1). Agregar 500 mL de HCl concentrado a 400 mL de agua contenidos en un matraz volumétrico de 1 L y llevar al volumen. Disoluciones patrón estándar para cada analito. Estas disoluciones pueden ser compradas o preparadas a partir de productos químicos de ultra-alta pureza (99.99 % o de mayor pureza). Todas las sales deben ser secadas por 1 hora a 105°C, con algunas excepciones específicas anotadas. Muchas sales de metales son extremadamente tóxicas si son inhalados o ingeridos. Lavarse las manos cuidadosamente después de su manejo. Para la preparación de las disoluciones estándares concentradas típicas proceder conforme a lo siguiente: Calcular las concentraciones en base al peso de metal puro añadido, o mediante el uso de la fracción del elemento y el peso de la sal de metal añadido. Disolución concentrada de aluminio, 1 mL = 1 000 µg Al. En un vaso disolver 1.0 g of aluminio metálico con 4.0 mL de HCl (1:1) y 1.0 mL de HN03 conc. Calentar lentamente para disolver el metal. Una vez que se haya disuelto completamente, transferir la disolución cuantitativamente a un matraz volumétrico de 1 000 mL, agregar 10.0 mL de HCl (1:1) y diluir al volumen con agua grado reactivo. Disolución concentrada de antimonio, 1 mL = 1 000 µg Sb. En un matraz volumétrico de 1 000 mL, disolver 2.667 3 g de K (SbO)C4H4O6 (fracción del elemento Sb = 0.374 9), con agua grado reactivo, adicionar 10 mL de HCl (1:1), y diluir al volumen con agua. Disolución concentrada de arsénico, 1 mL = 1000 µg As. En un matraz volumétrico de 1 000 mL disolver 1.320 3 g de As2O3 (fracción del elemento As = 0.757 4), con 100 mL de agua conteniendo 0.4 g de NaOH. Acidificar la disolución con 2 mL de HNO3 conc. y diluir al volumen con agua. Disolución concentrada de Bario, 1 mL = 1 000 µg de Ba. En un matraz volumétrico de 1 000 mL disolver 1.516 3 g de BaCl2 (fracción del elemento Ba = 0.659 5), previamente secado a 250 °C por 2 horas con 10 mL de agua y 1 mL de HCl (1:1). Adicionar 10 mL más de HCl (1:1) y diluir al volumen con agua. Disolución concentrada de Cadmio, 1 mL = 1 000 µg Cd. En un matraz volumétrico de 1 000 mL disolver 1.142 3 g de CdO (fracción del elemento Cd = 0.875 4), en una cantidad mínima de HNO3 (1:1). Calentar para incrementar la velocidad de disolución. Adicionar 10 mL de HNO3 concentrado y diluir al volumen con agua. Disolución concentrada de Cobre, 1 mL = 1 000 µg Cu. En un matraz volumétrico de 1 000 mL disolver 1.256 4 g de CuO (fracción del elemento Cu = 0.798 9) en una cantidad mínima de HNO3 (1:1). Adicionar 10 mL de HNO3 concentrado y diluir al volumen con agua. Disolución concentrada de Cromo, 1 mL = 1 000 µg Cr. En un matraz volumétrico de 1 000 mL disolver 1.923 1 g de CrO3 (fracción del elemento Cr = 0.520 0), en agua. Cuando se haya disuelto acidificar con 10 mL de HNO3 concentrado y diluir al volumen con agua. Disolución concentrada de Fierro (1 000 µg/mL Fe) - En un matraz volumétrico de 1 000 mL disolver 1.429 8 g de Fe2O3 en una mezcla caliente de 20 mL de HCl al 50 % y 2 mL de HNO3 concentrado. Diluir al volumen con agua. Disolución concentrada de Itrio, 1 mL = 1 000 µg Y. En un matraz volumétrico de 1 000 mL disolver 4.308 1 g de Y(NO3)3.6H20 (fracción del elemento Y = 0.232 1), en una cantidad mínima de HNO3 diluido. Adicionar 10 mL de HNO3 concentrado y llevar al volumen con agua. Disolución concentrada de Manganeso, 1 mL = 1 000 µg Mn. En un matraz volumétrico de 1 000 mL disolver 1.0 g de manganeso metálico, en una mezcla ácida (10 mL de HCl concentrado y 1 mL de HNO3 concentrado) y llevar al volumen con agua. Disolución concentrada de Níquel, 1 mL = 1 000 µg Ni. En un matraz volumétrico de 1 000 mL disolver 1.0 g de níquel metálico en 10 mL de HNO3 concentrado caliente, y llevar al volumen con agua. Disolución concentrada de Plata, 1 mL = 1 000 µg Ag. En un matraz volumétrico de 1 000 mL, disolver 1.574 8 g AgNO3 (fracción del elemento Ag = 0.635 0), agua, agregar 10 mL de HNO3 concentrado y llevar al volumen con agua. Disolución concentrada de Plomo, 1 mL = 1 000 µg Pb. En un matraz volumétrico de 1 000 mL disolver 1.598 5 g de Pb(NO3)2 (fracción del elemento Pb = 0.625 6), en una cantidad mínima de HNO3 (1:1). Adicionar 10 mL más de HNO3 (1:1) y diluir al volumen con agua. Disolución concentrada de Selenio, 1 mL = 1 000 µg Se. No secar. En un matraz volumétrico de 1 000 mL disolver 1.633 2 g de H2SeO3 (fracción del elemento Se = 0.612 3) en agua, y llevar al volumen con agua. Soluciones multielemento, con trazabilidad a NIST comercialmente disponibles, pueden utilizarse para preparar la curva de calibración. B.4.2.6 Procedimiento B.4.2.6.1 Preparación de las muestras Es la responsabilidad del laboratorio cumplir con todas las disposiciones aplicables en materia de manejo de residuos, particularmente las reglas de identificación, almacenamiento y disposición de residuos peligrosos. Asegurarse que todo el material utilizado durante el análisis esté libre de contaminación mediante procedimientos de lavado y descontaminación con ácido. Para descontaminar los vasos de digestión se puede utilizar un programa de digestión con el horno de microondas siguiendo las instrucciones del manual del fabricante. Si la muestra es incolora, transparente y no presenta turbiedad aparente, no se requiere realizar digestión, sólo adicionar un volumen apropiado de ácido nítrico para ajustar la concentración del ácido al 1% (v/v). Las muestras de control de calidad (ejemplo blanco de calibración, blanco de reactivo), deben someterse a los mismos procedimientos que las muestras incluyendo la adición del estándar interno o igualar la matriz con las disoluciones estándar. En caso de que la muestra presente turbiedad, llevar a cabo la digestión de la muestra. Medir una alícuota de 45 mL de muestra homogénea (AGITAR VIGOROSAMENTE LA MUESTRA), y depositar dentro de los vasos de digestión. Tomar 25 mL ó 10 mL de muestra + 20 mL de agua tipo I en caso de que la muestra presente materia orgánica o sólidos suspendidos. Adicionar 4 mL de HNO3 y 1 mL de HCl a cada vaso, tapar y colocar los vasos en el carrusel del horno de microondas. Proceder con la digestión de las muestras utilizando la potencia, rampas de temperatura y tiempo de acuerdo a las recomendaciones del proveedor. Preparar un blanco de reactivos utilizando agua tipo I en lugar de muestra y proceder de la misma manera. Una vez realizada la digestión permitir que los vasos se enfríen aproximadamente 5 minutos antes de sacar el carrusel del horno, posteriormente sacar el carrusel del horno y permitir que los vasos alcancen la temperatura ambiente (de 30 a 40 min) antes de destaparlos. Destapar cuidadosamente cada vaso dentro de la campana de extracción. Llevar la muestra a un volumen final de 50 mL, si la muestra digerida contiene partículas las cuales puedan obstruir el nebulizador, filtrar utilizando papel filtro Número. 2, 40 ó 41, posteriormente transferir a un tubo previamente etiquetado. B.4.2.6.2 Análisis de las muestras en el ICP-OES Calibrar el instrumento de acuerdo con los procedimientos recomendados por el fabricante, utilizando la mezcla de disoluciones estándar de calibración. Enjuagar el sistema con el blanco de calibración entre cada estándar o como recomienda el fabricante. Enjuagar el sistema con la disolución blanco de calibración, leer el CVI, el blanco de reactivos y las muestras. Enjuagar el sistema con la disolución blanco de calibración antes del análisis de cada muestra. El tiempo de lavado debe ser un minuto. Una reducción de este tiempo de enjuague deberá demostrarse adecuadamente. Registrar los resultados. Cuando la calibración inicial se realiza utilizando un solo nivel alto y el blanco de calibración, además del CVI, se debe analizar una disolución estándar de verificación de calibración continua de bajo nivel (low-level continuing calibration verification standard, LLCCV) antes del análisis de las muestras. Analizar después de cada diez muestras y al final del lote de análisis una disolución estándar de verificación de calibración continua (CCC) y un blanco de calibración continua (continuing calibration blank, CCB). B.4.2.6.3 Análisis de datos y cálculos Reportar directamente los datos generados del instrumento con su factor de dilución en caso de que exista considerando lo siguiente: Cuando se tenga un %DER mayor del 10 %, pero éste se deba a una lectura errónea de las 3 réplicas realizadas por el equipo, verificar si esa lectura se debió a un error del equipo, en cuyo caso considerar las otras 2 lecturas y evaluar si los resultados son válidos para ser reportados. Cuando se tenga un %DER menor del 10 % y el valor de la concentración reportada sea mayor que el límite de detección del método (LDM), verificar que el pico de respuesta del equipo corresponde al metal en cuestión, en cuyo caso se debe reportar la concentración obtenida, de lo contrario reportar como "no detectado", pues se estaría reportando un falso positivo. Para esto compare el pico del metal en cuestión sobreponiendo el pico correspondiente del estándar de calibración de concentración más cercana. B.4.2.6.4 Informe de prueba Reportar los resultados de las muestras en mg/L B.4.2.7 Calibración e interferencias B.4.2.7.1 Calibración B.4.2.7.1.1 Mezcla de disoluciones estándar de calibración Preparar una mezcla de disoluciones estándar de calibración de acuerdo con la Tabla B.4.2.-1., mediante la combinación de volúmenes apropiados de las soluciones concentradas. Tener cuidado al preparar la mezcla de estándares para asegurar que los elementos son compatibles y estables juntos. Transferir las disoluciones estándar a envases de fluorocarbono (FEP) o envases de polietileno o polipropileno que no hayan sido utilizados previamente para almacenar. Para todas las disoluciones estándar intermedios y de trabajo, especialmente estándares con una concentración baja (<1 mg/L o ppm), debe demostrarse su estabilidad antes de su uso. Preparar la mezcla de estándares con la periodicidad requerida, tomando en cuenta que la concentración puede cambiar con el tiempo. En la Tabla B.4.2-1 se muestran algunas combinaciones típicas de mezclas de disoluciones estándar de calibración.
Si al adicionar la plata, se forma inicialmente un precipitado, añadir 15 mL de agua y calentar el matraz hasta que la disolución se torne clara, la concentración de plata debe limitarse a 2 mg/L. La plata es estable en estas condiciones en una matriz de agua durante 30 días, si está protegida de la luz. Concentraciones más altas de plata requieren HCl adicional. Para preparar el blanco de calibración debe de acidificarse el agua a la misma concentración que los ácidos utilizados en los estándares de calibración. Los blancos de reactivos de laboratorio, deben contener todos los reactivos en los mismos volúmenes que se utilizan para las muestras y ser expuesto al proceso completo de preparación de las muestras incluyendo la digestión cuando es requerida. En el caso de la disolución estándar de verificación de la calibración inicial (CVI), preparar con las mismas mezclas de ácidos que los estándares de calibración. El CVI debe provenir de una fuente externa y diferente a los estándares de calibración y a una concentración cercana al punto medio de la curva de calibración. El CVI se requiere para la operación inicial y periódica de la verificación de los estándares de calibración y monitorear el desempeño del instrumento y el proceso analítico. Para la preparación de la disolución de verificación de calibración continua (CCC), preparar concentraciones conocidas de elementos que proporcionan interferencia que servirán para obtener los factores de corrección de interferencia, particularmente aquellos con interferencias conocidas en concentraciones de 0.5 a 1 mg/L. En ausencia del analito a medir, la sobrecorrección puede dar un valor negativo que puede ser reportado como cero. Si el instrumento en particular con la sobrecorrección da un valor negativo, no será necesaria la fortificación. Una forma adicional de preparar la curva de calibración puede ser empleando soluciones multi elemento con trazabilidad a NIST comercialmente disponibles, por ejemplo: SM-148-009 "ICP SOLUTION STOCK" SOLUCION A (High Purity Standards) que contiene Arsénico, Bario, Berilio, Boro, Bismuto, Fierro, Plomo, Litio, Manganeso, Selenio, Estroncio y Talio en una concentración de 1000 µg/mL±0.5% en HNO3 + Tr. HF con una densidad 1.098 g/cm3 ó mezcla analítica SM-148-009 (High-Purity Standards) que contiene Plata, Cadmio, Cromo, Cobalto, Cubre, Molibdeno, Níquel, Estaño, Silicio, Antimonio, Titanio, Vanadio y Zinc en una concentración de 1000µg/mL±0.5% en HNO3 con una densidad 1.084 g/cm3. Las referencias a marcas específicas y números de catálogo se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden representar especificaciones adecuadas para los artículos. Es recomendable preparar la curva de calibración de forma gravimétrica considerando las densidades correspondientes de la solución concentrada o stock para realizar el cálculo correspondiente de la concentración. Para determinar la concentración de las soluciones estándar si se preparan de forma gravimétrica debe utilizarse la ecuación siguiente: 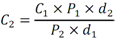 En donde: C2 concentración final mg/L C1 concentración inicial mg/L P1 masa del patrón de referencia a diluir en gramos d2 densidad del disolvente, HNO3 al 5% (V/V) P2 masa final de la disolución, aforo en gramos d1 densidad del soluto, material de referencia B.4.2.7.1.2 Optimización del instrumento Cargar el instrumento con los parámetros de funcionamiento adecuados establecidos como se detalla a continuación. Permitir que el instrumento se estabilice térmicamente antes de comenzar con la calibración (generalmente un mínimo de 30 minutos de funcionamiento). Operar el equipo de acuerdo a las instrucciones proporcionadas por el fabricante. La sensibilidad y los intervalos de trabajo para cada elemento variarán con la longitud de onda, espectrómetro, matriz y condiciones de operación. Consulte las instrucciones del fabricante para las longitudes de onda analíticas recomendadas y los límites de detección del instrumental (LDI). Antes de utilizar este procedimiento para el análisis de rutina, se deben realizar las pruebas iniciales de desempeño los cuales deben estar documentados y deben incluir los criterios de selección de los puntos de corrección de fondo; los intervalos de análisis, las ecuaciones aplicables, así como los límites superiores de estos intervalos; los límites de detección del método y del instrumento; y la determinación y verificación de las ecuaciones de corrección interelemento u otras rutinas de corrección de interferencias espectrales. Estos datos deben ser generados utilizando el mismo instrumento, condiciones de operación y la rutina de calibración que se utilizan para el análisis de muestras. Las condiciones de operación para soluciones acuosas usualmente varían de acuerdo a lo siguiente: · 1100 - 1200 watts de potencia, · 14 - 18 mm de altura, · 15 - 19 L/min de flujo de argón, · 0.6 - 1.5 L/min flujo de argón en el nebulizador, · 1 - 1.8 mL/min de velocidad de bombeo de la muestra con un tiempo de pre-limpieza de 1 minuto y el tiempo de medición aproximado de 1 segundo por pico de longitud de onda para instrumentos secuenciales y 10 segundos por muestra con instrumentos simultáneos. Para un plasma axial, las condiciones usualmente varían de acuerdo a lo siguiente: · 1100 a 1500 watts de potencia, · 15 a 19 L/min de flujo de argón refrigerante, · 0.6 a 1.5 L/min de caudal de flujo de argón nebulizador, · 1 a 1.8 mL de velocidad de bombeo de la muestra/min con un tiempo de pre-limpieza de 1 minuto y tiempo de medición cerca de 1 segundo por pico de longitud de onda para instrumentos secuenciales y 10 segundos por muestra para instrumentos simultáneos. Para alcanzar factores de corrección de interferencia repetibles se recomienda ajustar el flujo de argón aerosol para reproducir la relación de intensidad de Cu/Mn en 324.754 nm y 257.610 nm, respectivamente. B.4.2.7.1.3 Optimización del plasma Optimizar las condiciones de funcionamiento del plasma antes del uso del instrumento. El objetivo de la optimización de plasma es proporcionar un máximo de la señal de fondo de algunos de los elementos menos sensibles en la matriz de análisis. El uso de un controlador de flujo de masa para regular el flujo de gas nebulizador o una fuente de optimización de software facilita en gran medida el procedimiento. No es necesario llevar a cabo esta rutina diariamente, sólo se requiere cuando se ajusta por primera vez un instrumento nuevo, o después de un cambio en las condiciones de funcionamiento. Para la optimización del instrumento se recomienda realizar lo siguiente, o seguir las recomendaciones del fabricante. Encender el plasma radial y seleccionar una apropiada potencia de RF incidente. Dejar que el instrumento se encuentre térmicamente estable (entre 30 a 60 minutos) antes de comenzar. Mientras se aspira una disolución de 1 000 µg/L de itrio, seguir las instrucciones del fabricante del instrumento y ajustar el flujo del gas acarreador de aerosol a través del nebulizador hasta que una región de emisión azul definitiva del plasma se extienda aproximadamente de 5 a 20 mm por encima de la parte superior de la bobina de carga. Registrar el flujo del gas nebulizador y el ajuste de presión para futuras referencias. La solución de itrio también se puede utilizar para la alineación óptica gruesa de la antorcha mediante la observación de la superposición de la luz azul sobre la rendija (slit) de entrada hacia el sistema óptico. Después de establecer el flujo del gas nebulizador, determinar la velocidad de absorción de solución del nebulizador en mL/min mediante la aspiración de un volumen conocido de un blanco de calibración por un período de al menos tres minutos. Dividir el volumen aspirado por el tiempo en minutos y registrar la velocidad de absorción. Colocar la bomba peristáltica para entregar esa velocidad en un flujo constante. Calibrar el instrumento para alinearlo ópticamente, como será utilizado durante el análisis. El siguiente procedimiento se puede usar tanto para la optimización horizontal y vertical en el modo radial, pero está descrito para el vertical. Aspirar una disolución que contenga 10 µg/L de varios elementos seleccionados. As, Se, y Pb son los menos sensibles de los elementos y con mayor necesidad de optimización. Sin embargo, otros elementos pueden ser utilizados (Cr, Cu, Li y Mn también se pueden utilizar con éxito). Recolectar datos de intensidad en el pico de longitud de onda para cada analito a intervalos de 1 mm de 14 a 18 mm por encima de la bobina de carga. (Esta región del plasma es referida como la zona analítica). Repetir el procedimiento utilizando el blanco de calibración. Determinar la señal neta a la intensidad de blanco para cada analito para cada ajuste de altura de observación. Elegir la altura para ver el plasma que ofrece las mejores relaciones de intensidad (ratios) neta de los elementos analizados o la relación de intensidad más alta para el elemento de menor sensibilidad. Para la optimización en el modo axial, seguir las instrucciones del fabricante del instrumento. Las condiciones de operación óptimas del instrumento deben proporcionar los límites de detección del instrumental (LDI) más bajos. Si se cambian las condiciones de operación del instrumento, tales como la energía incidente o flujo de gas nebulizador, o si se instala un tubo inyector nuevo de la antorcha con un orificio de diámetro interno diferente, el plasma y la altura de visión deben ser re-optimizados. Después de completar la optimización inicial de operación de las condiciones de funcionamiento, y antes de analizar las muestras, el laboratorio debe establecer y verificar inicialmente una rutina de corrección de interferencias espectrales interelemento para ser utilizado durante el análisis de la muestra. El criterio para determinar que una interferencia espectral interelemento está presente, es una concentración positiva o negativa aparente para el analito que está más allá de un ± del límite establecido cercano a cero. Una vez establecida, la rutina completa debe ser verificada cada seis meses. Sólo una parte de la rutina de corrección se debe verificar con mayor frecuencia o cada vez que se llevan a cabo los análisis. Se deben mantener los registros de las verificaciones iniciales y periódicas. Antes de la calibración diaria, y después del período de calentamiento del instrumento, se tiene que optimizar el flujo del gas nebulizador. Si se utiliza un controlador de flujo de masa, se debe ajustar a la velocidad de flujo optimizado registrado. A fin de mantener válidas las rutinas de corrección interelemento espectrales, el flujo de gas nebulizador deberá ser el mismo (el cambio debe ser <2 %) día a día. Para la operación con disolventes orgánicos, se recomienda el uso de la entrada de argón auxiliar así como tubería resistente a los disolventes, el aumento de flujo de argón, la disminución del flujo del nebulizador, y el aumento de potencia de RF, para obtener un funcionamiento estable y mediciones precisas. B.4.2.7.1.4 Optimización del método Establecer el intervalo lineal para cada longitud de onda utilizada mediante la determinación de la señal de respuesta a partir de un mínimo de tres, preferentemente cinco, diferentes patrones de concentración en todo el intervalo. Cada vez que haya un cambio significativo en la respuesta del instrumento se debe determinar nuevamente el intervalo. Como mínimo, el intervalo debe ser revisado cada seis meses. Muchos de los metales alcalinos y alcalinotérreos tienen curvas de respuesta no lineales debido a la ionización y a los efectos de auto-absorción. Estas curvas se pueden utilizar si el instrumento lo permite; sin embargo, el intervalo efectivo debe ser revisado y el ajuste de la curva de segundo orden debe tener un coeficiente de correlación > 0.995. Estas respuestas de curvas no lineales deben ser revalidadas y se deben calcular cada seis meses. Estas curvas son mucho más sensibles a los cambios en las condiciones de operación que las lineales y debe realizarse cada vez que se han producido cambios moderados en el equipo. Establecer la sensibilidad, el límite de detección del instrumental (LDI), la precisión, el intervalo lineal, y los efectos de interferencia para cada analito individual de cada instrumento en particular. Todas las mediciones deben estar dentro del intervalo lineal del instrumento donde las ecuaciones de corrección son válidas. El laboratorio debe establecer el límite de cuantificación en el nivel bajo del intervalo para cada analito, en la mayoría de los casos es el punto más bajo de la curva de calibración, la verificación inicial se lleva a cabo utilizando 7 replicados de la muestra fortificadas en el nivel bajo y procesadas a través de todas las etapas de preparación y análisis del método. La recuperación media debe ser +/- 35% del valor real y la DER debe ser <20%. La verificación continua del el límite de cuantificación en el nivel bajo del intervalo, como mínimo se realiza trimestralmente para validar la capacidad de cuantificación a bajos niveles de concentración del analito. Para poder utilizar este método el laboratorio debe demostrar que el intervalo de trabajo para cada analito contempla la especificación establecida por las normas oficiales mexicanas correspondientes. B.4.2.7.1.5 Calibración diaria del instrumento Los estándares de calibración deben prepararse utilizando el mismo tipo de ácido o combinación de ácidos y en la misma concentración que las muestras. Preparar un blanco de calibración y mínimo 3 niveles de concentración para cada uno de los analitos a cuantificar. La concentración más alta no debe ser superior al intervalo lineal del instrumento según lo establecido anteriormente. Calibrar el instrumento diariamente y el coeficiente de correlación debe ser 0.995. La calibración inicial puede también realizarse utilizando un blanco de calibración y un solo nivel de concentración (nivel alto); si se realiza de esta manera, la curva resultante debe verificarse con dos disoluciones estándar (niveles bajo y medio del intervalo). El intervalo de aceptación de ambas disoluciones es del 80 a 120%. Comprobar la estandarización del instrumento mediante el análisis apropiado de muestras de control de calidad (MCC) de la siguiente manera: Preparar los estándares de calibración cada vez que se analiza una serie de muestras. Si la CVI se prepara diariamente y los resultados de los análisis del CVI están dentro de los criterios de aceptación, se puede ampliar la vigencia de las disoluciones mientras la disolución CVI cumpla con los criterios establecidos. El valor absoluto de los resultados del blanco de calibración debe ser menor que el límite de cuantificación en el nivel bajo del intervalo de la muestra de verificación para cada analito, o debe ser menor que el nivel de contaminación aceptable en el blanco especificado durante la validación del método. Si este no es el caso, debe ser identificada y corregida la razón de la condición fuera de control, y las diez muestras anteriores se deben volver a analizar. Preparar una disolución estándar a una concentración cercana al límite superior. El valor calculado debe estar dentro ± 10 % del valor real. Después de la calibración inicial, verificar analizando el CVI, esta disolución debe ser preparada a partir de un material independiente (segunda fuente) en o cerca del nivel medio de la curva de calibración. El criterio de aceptación para el CVI es de ± 10% de su valor real, Si no se cumple este criterio, se debe determinar la causa y volver a calibrar el instrumento antes del análisis de las muestras. Mantener registros de los resultados obtenidos del CVI junto con las muestras. La curva de calibración debe ser verificada al final de cada lote de análisis y después de cada 10 muestras mediante el uso de una disolución estándar de verificación de la calibración continua (CCC) y un blanco de calibración continua (CCB). La CCC debe prepararse a partir del mismo material que el CVI en o cerca del nivel medio de la curva de calibración. El criterio de aceptación para el estándar CCC deben ser de ± 10% de su valor real, y el CCB no debe contener analitos de interés mayores a 2 a 3 veces el límite de cuantificación en el nivel bajo del intervalo. Si los resultados obtenidos no se encuentran dentro de los límites especificados, interrumpir el análisis de la muestra, determinar la causa y volver a calibrar el instrumento. Posteriormente, volver a analizar todas las muestras que se corrieron después de la última CCB y CCC aceptables. Mantener registros de los resultados obtenidos junto con las muestras. Si se realiza la curva de calibración inicial con un solo estándar de calibración y un blanco, también se debe verificar antes del análisis de las muestras con una disolución estándar de verificación de la calibración de nivel bajo (LLCCV). La disolución estándar LLCCV debe prepararse a partir del mismo material que el CVI en el límite de cuantificación obtenido durante la validación. El criterio de aceptación para el LLCCV es de ± 20% de su valor real. Si el resultado no se encuentra dentro del límite especificado, el análisis de la muestra no puede comenzar hasta que se determine la causa y el resultado del LLCCV sea satisfactorio. El instrumento puede necesitar ser recalibrado o el límite de cuantificación ajustado. Mantener registros de los resultados obtenidos junto con las muestras. Diluir las muestras cuyo valor esté por encima del nivel alto de la curva de calibración y volver a analizar. B.4.2.7.2 Interferencias Las interferencias espectrales son causadas por emisión de fenómenos continuos o de recombinación, la luz extraviada de una línea de emisión de un elemento a alta concentración, la sobreposición de líneas espectrales de otro elemento o por sobreposición de espectros de banda moleculares no resueltas. La emisión de ruido de fondo de luz extraviada puede compensarse generalmente por la substracción de la emisión de fondo medida a los lados adyacentes del pico de la longitud de onda del elemento. Los barridos espectrales de las muestras o de soluciones unielementales en las regiones analíticas puede indicar cuándo es conveniente utilizar una longitud de onda alterna por alguna interferencia espectral, además, puede mostrar las posiciones más apropiadas de corrección de fondo a los lados adyacentes del pico del elemento o a un solo lado. La localización de la posición para medir la intensidad de ruido de fondo va a ser determinada por la complejidad del espectro adyacente al pico de la longitud de onda del analito. Estas localizaciones deben estar libres de interferencias espectrales (interelemento o moleculares), o adecuadamente corregidas para reflejar el mismo cambio en la intensidad de fondo como ocurre en el pico de la longitud de onda. Las sobreposiciones espectrales pueden evitarse utilizando una longitud de onda alterna o pueden compensarse por ecuaciones que corrigen contribuciones por interferencias interelemento, los instrumentos que utilizan ecuaciones para la corrección interelemento requieren que sean analizados los elementos interferentes al mismo tiempo que los analitos de interés. Cuando estas interferencias no son corregidas, generarán determinaciones falsas positivas o parcialmente positivas. El analista puede aplicar factores de corrección interelemento calculados en su instrumento con los intervalos de concentración probados para compensar los efectos de elementos interferentes. En la Tabla B.4.2-2, de este Apéndice, se muestran algunas interferencias espectrales (IEC) potenciales observadas para las longitudes de onda recomendadas. Cuando se usan ecuaciones de corrección interelemento, la interferencia puede expresarse como equivalentes de concentración del analito (es decir, concentraciones de analito falsas positivas) que resultan de 100 mg/L del elemento de interferencia. Por ejemplo, si el As se determina a 193.696 nm en una muestra que contiene aproximadamente 10 mg/L de Al, de acuerdo con la Tabla B.4.2-2, de este Apéndice, 100 mg/L de Al producirá una señal falsa positiva para un nivel de As equivalente a aproximadamente 1.3 mg/L. Por lo tanto, la presencia de 10 mg/L de Al resultará en una señal falsa positiva para As equivalente a aproximadamente 0.13 mg/L. Se advierte al usuario que otros instrumentos pueden presentar niveles de interferencia algo diferentes a los mostrados en la Tabla B.4.2-2, de este Apéndice. Los efectos de interferencia deben evaluarse para cada instrumento individual, ya que las intensidades variarán.
Las interferencias físicas están asociadas con la nebulización de la muestra y el proceso de transporte de la misma. Cambios en viscosidad y tensión superficial pueden causar malos resultados, especialmente en muestras conteniendo altas concentraciones de sólidos disueltos o altas concentraciones de ácido. Si existen interferencias físicas, estas deben ser reducidas: utilizando nebulizadores para un contenido alto de sólidos, diluyendo la muestra, utilizando una bomba peristáltica o utilizando un elemento apropiado como estándar interno. Otro problema que puede ocurrir con un alto contenido de sólidos disueltos es la acumulación de sales en el tubo inyector de la antorcha, lo cual afecta el flujo de transporte del aerosol ocasionando variación instrumental. Este problema puede controlarse utilizando: un nebulizador para un contenido alto de sólidos, humidificando el argón antes de la nebulización, lavando suficientemente entre muestra y muestra o diluyendo la muestra. Las interferencias químicas incluyen la formación de compuestos moleculares, efectos de ionización y efectos de vaporización del soluto. Normalmente, estos efectos no son significativos en la técnica de ICP-OES, pero si se observan pueden ser minimizadas mediante una cuidadosa selección de las condiciones de operación. Las interferencias de efecto de memoria son generadas cuando los analitos de una muestra anterior contribuyen en la medición de una nueva muestra. Los efectos de memoria pueden ocasionarse por la acumulación de la muestra en el tubo del nebulizador, en la antorcha del plasma y en la cámara de nebulización. Si se sospecha de interferencias por efectos de memoria, la muestra debe volver a analizarse después de un periodo prolongado de lavado. B.4.2.8 Seguridad Muchas sales de metales utilizadas para la preparación de las diluciones patrón estándar son extremadamente tóxicas si son inhalados o ingeridos. Lavarse las manos cuidadosamente después de su manejo. B.4.2.9 Referencias EPA. 2014. Method 6010D. Inductively coupled plasma-optical emission spectrometry. Rev. 4 B.5 MÉTODO DE PRUEBA PARA LA DETERMINACIÓN DE ÁCIDOS HALOACÉTICOS (ÁCIDO CLOROACÉTICO (MCAA); ÁCIDO DICLOROACÉTICO (DCAA); ÁCIDO TRICLOROACÉTICO (TCAA)) EN AGUA PARA USO Y CONSUMO HUMANO B.5.1 Definiciones y Términos Blanco de calibración, al volumen de agua grado reactivo que se utiliza para calibrar el instrumento. El blanco de calibración es un estándar cero. Blanco de campo, a la alícuota de agua grado reactivo que es colocada en un envase para muestra en el laboratorio, empacada para el muestreo y tratada como una muestra en todos los aspectos, incluyendo el contacto con los equipos de campo y expuesta a las condiciones del sitio de muestreo, almacenaje, preservación y todos los procedimientos analíticos, los cuales pueden incluir filtración. El propósito del blanco de campo es determinar si algún procedimiento de campo o transporte de la muestra ha contaminado la muestra. Blanco de reactivos, es una matriz libre de analitos a la cual se le agregan todos los reactivos en los mismos volúmenes o proporciones usadas en el procesamiento de la muestra. El blanco de reactivos debe llevarse a través de la preparación de la muestra y el procedimiento analítico. El blanco de reactivos se usa para documentar la contaminación resultante del proceso analítico. Desviación estándar, a la desviación estándar de la muestra (s), calculada a partir de n-1. No se refiere a la desviación estándar poblacional (s), calculada a partir de n. Estándar de calibración, a la solución preparada de un estándar diluido y/o una solución patrón y utilizada para calibrar la respuesta del instrumento con respecto a la concentración del analito. Estándar de verificación de la calibración, al punto medio del estándar de calibración que es utilizado para verificar la calibración inicial en el tiempo. Límite de detección del método (LDM), a la concentración mínima de un analito que puede identificarse, medirse y reportarse con una confianza del 99% cuando la concentración del analito es mayor a cero. Límite práctico de cuantificación (LPC), a la concentración mínima del analito que puede determinarse con un nivel de confianza predeterminado en condiciones rutinarias de operación. Este límite puede establecerse entre 5 a 10 veces el LDM. Matriz adicionada (MA) y Matriz adicionada duplicada (MAD), a la alícuota de una muestra ambiental para la cual cantidades conocidas de los avalitos del método son añadidas en el laboratorio. Las MA y MAD son analizados exactamente como una muestra. Su propósito es la cuantificación del sesgo y la precisión causada por la matriz de la muestra. Las concentraciones bases de los analitos en la matriz de la muestra debe determinarse en una alícuota separada y los valores medidos en las MA y MAD corregidas con las concentraciones base. Muestra de control de calidad (MCC), a la muestra sintética que contiene todos o un subgrupo de los analitos de interés del método a una concentración conocida. La MCC se obtiene de una fuente externa al laboratorio o es preparada de una fuente diferente de los estándares de la fuente de los estándares de calibración. Se usa para revisar el desempeño del laboratorio con materiales de prueba preparado externamente a los procesos normales de preparación. Rango de trabajo, al rango de la concentración sobre el cual la respuesta del instrumento para el analito es proporcional. B.5.2 Símbolos y términos abreviados CG/DCE cromatografía de gases con captura de electrones DCAA ácido dicloroacético DPR diferencia porcentual relativa %D diferencia porcentual LDM límite de detección del método LPC límite práctico de cuantificación MA matriz adicionada MAD matriz adicionada duplicada MCAA ácido cloroacético MTBE metil terbutil éter TCAA ácido tricloroacético B.5.3 Principio Se ajusta el pH a menos de 0.5 unidades de pH a un volumen medido de 40 mL de muestra de agua de uso y consumo humano y se extrae con 4mL de Metil Terbutil Éter (MTBE). Posteriormente se lleva a cabo la metilación con una solución ácida con metanol y calentamiento, el extracto ácido es neutralizado con una solución de bicarbonato de sodio y los analitos de interés son analizados y cuantificados con columna capilar y CG/CDE B.5.4 Alcance y aplicación El método descrito es un procedimiento para la determinación de ácidos haloacéticos (ácido cloroacético (MCAA); ácido dicloroacético (DCAA); ácido tricloroacético (TCAA) en agua de uso y consumo humano proveniente de sistemas de abastecimiento por cromatografía de gases con captura de electrones (CG/DCE). Este método debe ser utilizado sólo bajo la supervisión de analistas expertos en el uso de cromatografía de gases con captura de electrones CG/DCE y en la interpretación de sus resultados. Cada analista debe demostrar la habilidad para generar resultados aceptables con este método. Los métodos analíticos determinados experimentalmente para los ácidos haloacéticos se muestran en la Tabla B.5-1, de este Apéndice.
B.5.5 Equipos y materiales Las referencias a marcas específicas y números de catálogo se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden representar especificaciones adecuadas para los artículos. Balanza analítica. Con precisión de 0.0001 g Columna capilar cuantitativa DB 35 MS. De sílica fundida a 30 m de longitud por 0.25 mm con una fase estacionaria de 35% fenil metil silicón de 0.25 µm de grosor de película o equivalente. Columna capilar de confirmación DB-XLB. Capilar de sílica fundida de 30 m de longitud por 0.25 mm de diámetro interno con fase estacionaria de poliloxano y un grosor de película de 0.25 µm o equivalente. Cromatógrafo de gases (CG). Con inyector capilar y detector de captura de electrones (DCE) con sistema computarizado de registro y procesamiento de datos Matraz volumétrico tipo "A". De 4, 5 y 10 mL para la preparación de estándares. Microjeringa. De 10, 25, 50, 100 y 500 µL. Pipeta volumétrica tipo "A" de 4 mL. B.5.6 Reactivos y soluciones Los reactivos que requiere el método deben ser tipo ACS grado reactivo a menos que otra cosa se indique. Se deben preparar 5 niveles como mínimo de diferentes concentraciones de los analitos de interés para servir como disoluciones de calibración. Preparar los estándares de calibración de forma gravimétrica si no se cuenta con material calibrado. El almacenaje de las soluciones estándar (patrón, de calibración, interna y estándares surrogados) debe ser a una temperatura menor a 4°C en viales de 2 mL. Todas las soluciones estándares patrón deben reemplazarse después de un año o antes si la rutina de control de calidad indica degradación o evaporación del solvente. Acetona. Grado reactivo, nanogrado o equivalente. Bicarbonato de sodio. Grado reactivo. Cloruro de amonio. Grado reactivo. Disolución de 100 mg/L de metil cloroacetato, dimetilcloroacetato y 33.3 mg/L de metil tricloroacetato. La solución estándar puede prepararse de materiales estándares puros o puede ser una solución certificada. Partir de una solución estándar de Metil cloroacetato de 3 000 mg/L, metil dicloroacetato de 3 000 mg/L y metil tricloroacetato de 1 000 mg/L (en mezcla). En un matraz aforado de 1 mL poner 33.3 µL de la mezcla de metil cloroacetato, metil dicloroacetato y metil tricloroacetato, aforar con MTBE. Disolución de 1 y 0.33 mg/L de metil acetatos. La solución estándar puede prepararse de materiales estándares puros o puede ser una solución certificada. En un matraz aforado de 5 mL poner 50 µL de la mezcla de 100 mg/L de metil cloroacetato, 100 mg/L de metil dicloroacetato y 33.3 mg/L de metil tricloroacetato y aforar con MTBE. Esta disolución puede servir como estándar de calibración de 1 y 0.33 mg/L. Estándar de calibración de 0.01 mg/L de metil cloroacetato, metil dicloroacetato y 0.003 3 mg/L de metil tricloroacetato. En un matraz aforado de 1mL ponga 10 µL de la solución de 1 mg/L y aforar con MTBE. Los estándares de calibración, se pueden preparar gravimétricamente. Estándar de calibración de 0.02 mg/L de metil cloroacetato, metil dicloroacetato y 0.006 6 mg/L de metil tricloroacetato. En un matraz aforado de 1mL poner 20 µL de la solución de 1mg/L y aforar con MTBE. Los estándares de calibración, se pueden preparar gravimétricamente. Estándar de calibración de 0.05 mg/L de metil cloroacetato, metil dicloroacetato y 0.0166 mg/L de metil tricloroacetato. En un matraz aforado de 1mL poner 50 µL de la solución de 1 mg/L y aforar con MTBE. Los estándares de calibración, se pueden preparar gravimétricamente. Estándar de calibración de 0.1 mg/L de metil cloroacetato, metil dicloroacetato y 0.033 mg/L de metil tricloroacetato. En un matraz aforado de 1 mL poner 100 µL de la solución de 1 mg/L y aforar con MTBE. Los estándares de calibración, se pueden preparar gravimétricamente. Estándar de calibración de 0.2 mg/L de metil cloroacetato, metil dicloroacetato y 0.066 mg/L de metil tricloroacetato. En un matraz aforado de 1 mL poner 200 µL de la solución de 1mg/L y aforar con MTBE. Los estándares de calibración, se pueden preparar gravimétricamente. Estándar de calibración de 0.5 mg/L de metil cloroacetato, metil dicloroacetato y 0.166 mg/L de metil tricloroacetato. En un matraz aforado de 1 mL poner 500 µL de la solución de 1mg/L y aforar con MTBE. Los estándares de calibración, se pueden preparar gravimétricamente. Estándares internos. 1,2,3-tricloropropano. Solución concentrada de 25 mg. Estándares surrogados. 2,3 ácido dibromopropiónico. Solución concentrada de 1 000 mg/L. Metanol. Grado HPLC o equivalente. Metil terbutil éter. Grado HPLC o equivalente. Solución de ácido sulfúrico en Metanol al 10%. En un matraz aforado de 50 mL adicione de 20 a 30 mL posteriormente adicione lentamente 5 mL de ácido sulfúrico concentrado y afore a 50 mL con metanol. Solución de bicarbonato de sodio saturada. Adicionar el bicarbonato de sodio a un volumen de agua hasta saturación. Sulfato de cobre pentahidratado. Grado reactivo. Sulfato de sodio. Granular anhidro purificado a 600 °C durante 4 horas. Grado reactivo. B.5.7 Procedimiento B.5.7.1 Recolección, conservación y almacenamiento de muestras Es necesario contar con muestras de agua de uso y consumo humano a analizar con un volumen de 250 mL en envase de vidrio ámbar. No se requiere de ningún tratamiento especial en campo. Las muestras y los blancos de campo deberán preservarse con cloruro de amonio y refrigerarse a 4 °C hasta su análisis. Todas las muestras deben extraerse dentro de los 14 días después de la recolección, una vez extraídas se tiene un tiempo de 7 días si se preserva a 4 °C y 14 días si se preserva a -10 °C. B.5.7.2 Procedimiento analítico Extracción de la muestra. Tomar 40 mL de muestra y transferirlo a un vial de 60 mL, adicionar estándar surrogado y compuestos target (muestras fortificadas), adicionar por lo menos 2 mL de ácido sulfúrico concentrado para ajustar su pH menor a 0.5. Verificar con potenciómetro o con papel indicador de pH, rápidamente adicionar 2 g aproximadamente de sulfato cúprico pentahidratado, agitar hasta disolución, posteriormente adicionar rápidamente 16 g de sulfato de sodio y agitar de 3 a 5 minutos hasta que casi todo este disuelto, extraer con 4 mL de disolvente (MTBE), agitando vigorosamente durante 2 minutos. Dejar que se separen las fases durante 5 minutos. Metilación. Transferir aproximadamente 3 mL de la parte superior (MTBE) a un tubo de rosca de 15 cm con tapa y adicionar 1mL de ácido sulfúrico al 10% en metanol. Colocar el tubo en un Termoblock a 50 °C durante 2 horas. Pasado este tiempo dejar enfriar y adicionar 4mL de la solución saturada de bicarbonato de sodio; agitar por 2 minutos con cuidado de ventear el dióxido de carbono (CO2) generado por la reacción y transferir 1 mL del MTBE a un vial para su análisis cromatográfico. Las condiciones utilizadas para el análisis de extractos de muestras son las siguientes: Gas acarreador hidrógeno Presión en columna 9.4 psi Temperatura del inyector 200 °C Temperatura del detector 310 °C Temperatura inicial 40 °C Tiempo inicial 1 - 2.0 minutos Rampa 1 10 °C/minutos Temperatura final 1 80 °C Tiempo final 1 0 minutos Rampa 2 35 °C/minutos Temperatura final 2- 300 °C Tiempo final 2 0 minutos Inyección Split relación 10:1 En el procedimiento de análisis con el método de estándar interno, agregar el estándar al extracto de la muestra antes de aforar (opcional). Inyectar un volumen adecuado (1 a 3 µL) del extracto de la muestra o estándar en el cromatógrafo de gases. Registro el volumen final del extracto y el área del pico resultante. Identificar los ácidos haloacéticos en la muestra comparando los tiempos de retención de los picos en el cromatograma de muestra con los tiempos de retención de los picos en los cromatogramas de los estándares. Si la respuesta del pico sobrepasa el intervalo de concentración de la curva patrón, diluir el extracto y analizar nuevamente. La identificación de los ácidos haloacéticos utilizando en este método un detector de captura de electrones se basa en la concordancia entre los tiempos de retención de los picos en el cromatograma de la muestra con los intervalos de los tiempos de retención establecidos a través del análisis de los estándares. La identificación tentativa de un analito ocurre cuando un pico del extracto de una muestra cae dentro del intervalo de tiempo de retención establecido para un analito problema específico. Cada identificación tentativa debe confirmarse. La cuantificación de los ácidos haloacéticos, puede realizarse usando la técnica de estándar interno o estándar externo calculando la concentración. Puede usarse la confirmación por cromatografía de gases acoplada a espectrometría de masas (Gas chromatographymass spectrometry, GC/MS) en conjunción con el análisis siempre y cuando la concentración sea suficiente para su detección por CG/EM o bien por medio de una columna confirmatoria. El CG/EM debe calibrarse para los analitos problema. Cuando se usa una técnica SIM, los iones y los tiempos de retención deben ser característicos de los ácidos haloacéticos a ser confirmados. La confirmación de CD/EM debe completarse analizando los mismos extractos utilizados para el análisis CG/DCE y los extractos del blanco asociado. B.5.7.3 Análisis de datos y cálculos Para determinar la concentración de los ácidos haloacéticos, calcular la concentración en muestras líquidas mediante la siguiente ecuación: 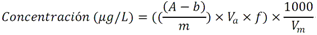 En donde: A área del parámetro a medir b ordenada al origen de la regresión lineal (de la curva de calibración) m pendiente de la regresión lineal (de la curva de calibración) Va volumen final del extracto en mL f factor de dilución Vm volumen de muestra extraída en mL B.5.7.4 Informe de prueba Reportar el resultado como µg/L de ácido cloroacético, ácido dicloroacético y ácido tricloroacético B.5.8 Control de calidad, calibración e interferencias B.5.8.1 Control de calidad B.5.8.1.1 Debe de llevarse a cabo una verificación del equipo. La verificación del detector de DCE debe realizarse de acuerdo con el manual de operación del equipo. Verificar que los flujos de gases y condiciones cromatográficas sean las adecuadas. Verificación de blanco electrónico. Realizar una corrida con las condiciones cromatográficas del método, pero sin inyectar nada, si el cromatograma presenta picos a cualquier tiempo de retención, se purga el sistema poniendo el horno a 300°C durante 15 minutos y se repite el análisis del blanco electrónico. B.5.8.1.2 Verificación de la calibración inicial. Deberá realizarse inyectando los estándares de calibración preparados y se considerará lineal si el % de la desviación estándar relativa (%RSD) de los factores de calibración (CF) es menor o igual al 20%, los cálculos se realizan con las siguientes ecuaciones: 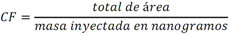 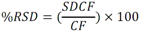 En donde: SDCF desviación estándar de los factores de calibración CF promedio de los factores de calibración La verificación de la calibración inicial puede también efectuarse mediante la regresión por mínimos cuadrados en la cual el criterio de aceptación es que el factor de correlación "r" sea mayor o igual a 0.99. Si los criterios para el %RSD o r no se cumplen se procede a la revisión del sistema cromatográfico y la preparación de las disoluciones de calibración. Posteriormente la curva de calibración será vigente, si el análisis de un punto intermedio de la curva no presenta una variación de la concentración mayor al 15% y que será calculada de la siguiente manera: 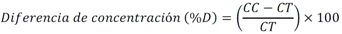 En donde: CC concentración calculada CT concentración teórica o si se utiliza el factor de calibración 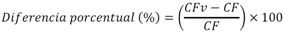 En donde: CFv factor de calibración de cada compuesto en el estándar de verificación CT factor de calibración del compuesto de la calibración inicial Si el criterio de aceptación para la diferencia porcentual es mayor al 15% se procederá a la revisión del sistema cromatográfico y a la preparación de la solución estándar utilizada. B.5.8.1.3 Verificación de contaminación de los reactivos. Los blancos de reactivos no deben presentar ningún tipo de contaminante al tiempo de retención de los ácidos haloacéticos medidos, por lo que es importante que los reactivos utilizados cumplan con las especificaciones mencionadas. Si los resultados de los blancos indican contaminación con picos a T.R. de los ácidos haloacéticos, ésta contaminación debe ser menor a los límites de regulación asociada a los ácidos haloacéticos o menor al 5% de los resultados de los mismos en las muestras. Si los resultados de los blancos no cumplen con los criterios mencionados debe localizar la fuente de contaminación y extraer y analizar nuevamente las muestras asociadas al blanco contaminado. Use también los blancos de reactivos para verificar la contaminación por arrastre de muestras con altas concentraciones en análisis secuenciales. B.5.8.1.4 Verificación del lote analítico. Por cada lote analítico se analice una muestra de control de calidad y se calcula el % de recuperación de cada analito y surrogados de la muestra control, los cuales deberán estar dentro del rango de 70 a 130%. De no ser así se revisará la preparación de la muestra control y el sistema cromatográfico. Realizar la corrección necesaria y reanalizar la muestra control. B.5.8.1.5 Verificación de interferencias de matriz. La verificación de interferencias por parte de la muestra se lleva a cabo mediante las muestras fortificadas. El laboratorio debe realizar el análisis de muestras fortificadas y muestras duplicadas, fortificar una muestra por cada lote. Las muestras fortificadas y muestras duplicadas sólo se analizarán cuando por las características de la muestra se sospeche de interferencias de matriz o donde sean parte de un plan de aseguramiento de calidad de un proyecto. Para determinar el efecto de la matriz en el recobro del analito. Debe analizar una muestra fortificada por cada lote de muestras. Calcular el % de recobro, el cual debe encontrarse dentro del 70 al 130%. Si los valores no cumplen con lo especificado, el sistema analítico está fuera de control, determine las causas y corrija el problema. Documente las incidencias y acciones correctivas. Para determinar el efecto de la matriz en la precisión del analito - precisión de la matriz duplicada. Analizar una muestra duplicada. Calcular la diferencia porcentual relativa, también llamada DPR, entre la muestra y se duplicada para cada analito, con la siguiente ecuación: 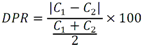 En donde: C1 concentración de la primera muestra C2 concentración de la segunda muestra (muestra duplicada) La DPR de cada analito obtenido entre la muestra y la duplicada debe ser <20%. Si los valores no cumplen con lo especificado, el sistema analítico está fuera de control, determine las causas y corrija el problema, documente las incidencias y acciones correctivas. Repita el análisis sólo del lote asociado con la muestra y la duplicada que provocó que la prueba del DPR fallara. B.5.8.1.6 Verificación de la estabilidad de la curva de calibración. En cada lote analizado, si más de 25 muestras van a ser analizadas durante el día, verificar la curva de calibración analizando un estándar de concentración media del intervalo de trabajo al principio del lote y después de cada 25 muestras ó 12 horas de análisis continuo. Calcule el factor de calibración o la concentración para cada ácido haloacético del estándar de concentración media y la diferencia porcentual (%D) con la siguiente ecuación: 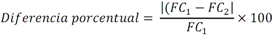 En donde: FC1 factor de calibración promedio para cada ácido haloacético en la curva de calibración, o la concentración nominal del estándar de verificación. FC2 factor de calibración promedio para cada ácido haloacético en el estándar de concentración media, o la concentración calculada en el estándar de verificación. El valor obtenido del % de la diferencia (%D) deberá ser menor al 15% de no ser así verificar la vigencia, revisar también el sistema cromatográfico y determinar la causa de la falla y corregir, de no lograr corregir la falla, recalibrar nuevamente y en el caso de que la falla del estándar de verificación sea en un lote mayor de 25 muestras reanalice las 10 muestras anteriores al estándar de verificación que fallo. B.5.8.1.7 Verificación de estándares surrogados. Evaluar los datos de recobro de los surrogados de las muestras. Calcular el % de recuperación de los estándares surrogados en blancos, muestras control, muestras fortificadas (si se realizaron), muestras duplicadas (si se realizaron) y muestras reales, con la siguiente ecuación: 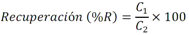 En donde: C1 concentración calculada de surrogados en la muestra C2 concentración nominal del surrogado adicionado a la muestra Si los valores no cumplen con los especificados, determinar las causas y corregir el problema, documentar las incidencias y acciones correctivas e incluirlas en el expediente del desarrollo inicial del desempeño del método. El %R deberá estar entre el 70% y el 130% de recuperación de no ser así verificar la preparación de la muestra que falló y el sistema cromatográfico, determinar la causa de la falla y corregir, de no encontrarla, repetir la extracción de la muestra y volver a analizar si el %R esta fuera del rango de aceptación, llenar hoja de incidencia y reportar como interferencia de matriz y los resultados obtenidos. B.5.8.1.8 Verificación de estándares internos (sólo para calibración de estándares internos). Compare el área de el o los estándares internos de las muestras con el área promedio de los estándares internos obtenidos de los puntos de calibración, el valor obtenido en el o los estándares internos de la muestra no deben ser menores a 50% ni mayores a 200% del valor promedio obtenido en la curva de calibración para cada estándar interno. Si los valores no cumplen con lo especificado, el sistema analítico está fuera de control, determinar las causas y corregir el problema, documentar las incidencias y acciones correctivas e incluirlas en el expediente del desarrollo inicial del desempeño del método. B.5.8.1.9 Composición del lote analítico (para cada 20 muestras): 1 blanco electrónico 2 muestra de verificación de la calibración continua 3 blanco de reactivos 4 muestra de control de calidad 5 muestras real no. 1 6 a 14 muestra real no. 2 a 10 15 muestra de verificación de la calibración continua 16 a 25 muestra real no.11 a 20 26 muestra duplicada (si se realizó) 27 muestra adicionada (si se realizó) 28 muestra de verificación de la calibración continua Las muestras de verificación de la calibración continua se correrán cada 10 muestras reales y al final de cada lote. B.5.8.1.10 Dependiendo de los requerimientos del programa específico de control de calidad, puede requerirse los duplicados de campo, para evaluar la precisión y la exactitud del muestreo y técnicas de transportación de la muestra. B.5.8.1.11 Especificaciones de aceptación y rechazo
B.5.8.2 Calibración Las condiciones de operación del cromatógrafo de gases están definidas por el tipo de columna elegida. Una vez establecidas las condiciones de operación inyecte un volumen adecuado (1 a 3 µL) de cada estándar de calibración. Otros volúmenes pueden ser utilizados si así lo requiere la sensibilidad de los compuestos de interés. Para llevar a cabo el procedimiento de calibración, analizar cada estándar de calibración a partir de las disoluciones. Si la técnica de cuantificación elegida es por estándar externo construir una curva de área o altura de pico contra concentración y verificar la linealidad de la curva. Si la DSR de cada ácido haloacético es £20%, entonces la respuesta del instrumento es considerada lineal y el factor de calibración promedio FC o FR puede utilizarse para la cuantificación de los resultados de las muestras. Si la DSR es mayor que 20%, entonces no puede asumirse linealidad. Si se utiliza regresión lineal por mínimos cuadrados, deberá obtener el factor de correlación "r" para cada analito, el cual deberá ser mayor o igual a 0.99 si "r" es menor a este valor, la curva de calibración no es lineal y deberá verificarse la preparación de los estándares de calibración y el sistema cromatográfico y repetir la calibración. Si el valor del FR sobre el rango de trabajo es constante, es decir, menor que el 20% de la desviación estándar relativa, el FR puede tomarse como invariable y el promedio del FR puede utilizarse para los cálculos, Alternativamente, los resultados pueden utilizarse para graficar una curva de calibración de los promedios de la respuesta contra FR. La curva de calibración o FR deben verificarse en cada lote de análisis, por medio de la medición de uno más estándares de calibración. Si la respuesta para cualquier parámetro varía de la respuesta predicha por más de ±15%, la prueba debe repetirse utilizando un estándar de calibración nuevo. Alternativamente, deberá prepararse una nueva curva de calibración para cada parámetro. Debe verificarse la estabilidad de la curva de calibración. En cada lote analizado durante el día, la curva de calibración debe verificarse con un estándar de concentración media del rango de trabajo al principio del lote, después de cada 10 muestras y al final de cada lote de análisis. El valor DR de la muestra debe estar dentro del 15% del valor de la calibración inicial, de no ser así entonces debe identificar el problema, corregir y repetirse la verificación de la calibración. B.5.8.3 Interferencias Las interferencias co-extraídas con la muestra variarán considerablemente de matriz a matriz. Mientras las técnicas de limpieza generales se proporcionan como parte de este método, algunas muestras deben requerir de una limpieza adicional para obtener la ejecución de un grado de discriminación y cuantificación. Las fuentes de interferencias en este método pueden ser debidas, entre otras, las siguientes: Las interferencias de matriz, son los compuestos extraídos de las muestras y con las que el detector tiene alguna respuesta, para eliminar este tipo de interferencias se utilizan los procedimientos de limpieza de los extractos adecuados. Las interferencias instrumentales, son aquellas ocasionadas por las impurezas del gas de acarreo o por fallas del instrumento, para la cual se verifican blancos electrónicos y procedimientos de limpieza térmica. Las interferencias por ftalatos introducidos durante la preparación de las muestras pueden dar un problema mayor en la determinación de los haloacéticos. Los contenedores comunes de plástico varían en la cantidad de ftalatos los cuales son extraídos fácilmente o lixiviados de los mismos materiales durante las operaciones del laboratorio. Las interferencias de ftalatos pueden disminuirse de mejor forma, impidiendo el contacto con cualquier material plástico y revisando todos los solventes y reactivos por contaminación de ftalatos. Para eliminar una contaminación de ftalatos, se requiere de una limpieza exhaustiva de los solventes reactivos y material de vidrio. Estos materiales pueden removerse a través del uso del método de limpieza con ácido sulfúrico/permanganato. Los límites de detección en las muestras reales dependerán de las interferencias presentes en las mismas por lo que es muy importante tener en cuenta que pueden variar hasta en magnitudes de 10 a 1000 veces (factores de concentración de las muestras). B.5.9 Seguridad, prevención de contaminación y manejo de residuos Cada reactivo utilizado en estos procedimientos debe ser tratado como un riesgo potencial para la salud y la exposición a estos materiales debe ser minimizada. Cada laboratorio es responsable de mantener un conocimiento de las regulaciones respecto a la manipulación segura de cualquier producto químico usado en este método. Debe ponerse a disposición de todo el personal involucrado en el análisis las hojas de datos de seguridad de los productos químicos. Debe de utilizarse equipo de protección personal apropiado para el manejo de muestras y estándares. Los estándares primarios de los ácidos haloacéticos deberán prepararse en una campana. Cuando el analista maneje altas concentraciones de compuestos tóxicos deberá utilizarse un respirador de gases tóxicos. Es responsabilidad del laboratorio cumplir con todos los reglamentos federales, estatales y locales referentes al manejo de residuos, particularmente las reglas de identificación, almacenamiento y disposición de residuos peligrosos. B.5.10 Referencias EPA. 1995. Method 552.2 Determination of Haloacetics Acids and Dalapon in Drinking Water by Liquid-Liquid Extraction, Derivatization and Gas Chromatography with Electron Capture Detector. Rev. 1.0 B.6 MÉTODO DE PRUEBA PARA LA DETERMINACIÓN DE BROMATO, CLORATO Y CLORITO EN AGUA PARA USO Y CONSUMO HUMANO B.6.1 Definiciones y términos Blanco de campo, a la alícuota de agua grado reactivo que es colocada en un envase para muestra en el laboratorio, empacada para el muestreo y tratada como una muestra en todos los aspectos, incluyendo el contacto con los equipos de campo y expuesta a las condiciones del sitio de muestreo, almacenaje, preservación y todos los procedimientos analíticos, los cuales pueden incluir filtración. El propósito del blanco de campo es determinar cuál procedimiento de campo o transporte de muestra y ambiente ha contaminado la muestra. Desviación estándar, a la desviación estándar de la muestra (s), calculada a partir de n-1. Disolución madre, corresponde a la disolución de máxima concentración en un análisis. Es a partir de esta disolución que se preparan las disoluciones de trabajo. Disolución patrón, a la disolución de concentración conocida preparada a partir de un patrón primario. Estándar de calibración, a la solución preparada de un estándar diluido y/o una solución patrón y utilizada para calibrar la respuesta del instrumento con respecto a la concentración del analito. Estándar de verificación de la calibración, punto medio del estándar de calibración que es utilizado para verificar la calibración inicial en el tiempo. Intervalo de trabajo, intervalo de la concentración sobre la cual la respuesta del instrumento para el analito es proporcional. Matriz adicionada (MA) y Matriz adicionada duplicada (MAD), a la alícuota de una muestra ambiental para la cual cantidades conocidas de los analitos del método son añadidas en el laboratorio. Las MA y MAD son analizados exactamente como una muestra. Su propósito es la cuantificación del sesgo y la precisión causada por la matriz de la muestra. Las concentraciones bases de los analitos en la matriz de la muestra debe determinarse en una alícuota separada y los valores medidos en las MA y MAD corregidas con las concentraciones base. Muestra de control de calidad (MCC), a la muestra sintética que contiene todos o un subgrupo de los analitos de interés del método a una concentración conocida. La MCC se obtiene de una fuente externa al laboratorio o es preparada de una fuente diferente de los estándares de la fuente de los estándares de calibración. Se usa para revisar el desempeño del laboratorio con materiales de prueba preparado externamente a los procesos normales de preparación. Patrón primario, al patrón que es designado o reconocido ampliamente como un patrón que tiene las más altas cualidades metrológicas y cuyo valor es aceptado sin referencia a otros patrones de la misma magnitud. B.6.2 Símbolos y términos abreviados DSR desviación estándar relativa EDA etilendiamina µS microsiemens M matriz MA matriz adicionada MAD matriz adicionada duplicada MCC muestra de control de calidad MDL límite de detección del método MDUP matriz duplicada B.6.3 Principio La muestra es filtrada a través de un filtro de 0.45 µm en caso de presentar sedimentos o turbidez, posteriormente se le adicionan 50 µL de una disolución de EDA junto con el estándar surrogado y se analiza por la técnica de cromatografía de intercambio iónico con supresión. B.6.4 Alcance y aplicación Este método es utilizado para la determinación de aniones comunes e inorgánicos subproductos de la desinfección de agua por cromatografía de intercambio iónico y detector conductimétrico con supresor en muestras de agua para uso y consumo humano. Los aniones que pueden ser determinados a través de este método comprenden aniones inorgánicos subproductos de la desinfección (clorito, clorato, bromuro y bromato). El método descrito es un procedimiento para la determinación de bromato, bromuro, clorato y clorito por cromatografía iónica en agua para uso y consumo humano. B.6.5 Equipos y materiales Las referencias a marcas específicas y números de catálogo se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden representar especificaciones adecuadas para los artículos. Sólo se mencionan los equipos y materiales que no son de uso común en el laboratorio analítico. Todo el material volumétrico utilizado debe ser clase A o estar verificada su calibración. Balanza analítica. Con precisión de 0.0001 g Columna de intercambio aniónico, que cumpla con el desempeño requerido de éste método, por ejemplo con tamaño de partícula de 5 µm, 25 cm de longitud y 4.0 mm de diámetro interno o equivalente y guardacolumna 7.2 X 4.6 mm Cromatógrafo de intercambio iónico Detector conductimétrico, con un intervalo de trabajo de 0.01 a 5000 µS en 12 pasos, capaz de trabajar con polaridad positiva o negativa o equivalente. Filtros para jeringa de celulosa regenerada de 0.45µm de poro y 13mm de diámetro. De ser necesario use filtros de limpieza SPE-IC para eliminar interferencias de otros iones. Jeringa de vidrio o de plástico de 3 mL Matraz volumétrico de 1 L Microjeringas de 10, 50, 100 y 500 µL Pipeta volumétrica de 10 mL Supresor Termostato de columna (intervalo de temperatura ambiente a 60°C) B.6.6 Reactivos y soluciones Los reactivos que requiere el método deben ser tipo ACS grado reactivo a menos que otra cosa se indique. Agua grado HPLC. A menos que otra cosa se indique, el agua utilizada debe cumplir con las siguientes características: ser grado reactivo de acuerdo al estándar de la Sociedad Americana para Pruebas y Materiales tipo I (ASTM, American Society for Testing and Materials), libre de compuestos orgánicos y que no presente interferencia. El agua utilizada en la fase móvil debe ser filtrada a través de una membrana de 0.45 a 0.20 µm. Bromato 1000 mg/L material de referencia en solución Bicarbonato de sodio sal 100% o solución concentrada 0.5 M Clorato 1000 mg/L material de referencia en solución Clorito 1000 mg/L material de referencia en solución Dicloroacetato de Potasio sal (surrogado) Dilución 1000 mg/L de etilendiamina, diluir 6 µL de EDA en 2 mL de agua, mezclar y aforar a 5 mL con agua. Disolución 100 mg/mL de etilendiamina (preservador), diluir 2.5 mL de EDA en 10mL de agua y aforar a 25 mL con agua. Disolución de 50 mg/L EDA, esta solución se usa para diluir las muestras aforar todas las soluciones estándar y mezcla de verificación de aniones. Tomar 0.5 mL de la solución 1000 mg/L de EDA y aforar a 10 mL con agua. Etilendiamina (EDA) ensayo 99.9% o equivalente. B.6.7 Procedimiento B.6.7.1 Muestreo, preservación y manejo de muestra Tomar una muestra de mínimo 50 mL en un recipiente de plástico o vidrio ámbar. Sólo en caso de que la muestra contenga dióxido de cloro burbujear con helio o nitrógeno. Para preservar la muestra, adicionar 0.1 mL de EDA de 100 mg/mL, la concentración final es de 50 mg/L de EDA en la muestra. Las muestras deben mantenerse a una temperatura de 2 a 4°C desde el momento de su obtención hasta una hora antes del análisis. El tiempo límite para el análisis es de 14 días a partir de su recolección. B.6.7.2 Procedimiento analítico Para preparar la muestra, dejar que la muestra mantenida a baja temperatura se encuentra a temperatura ambiente antes del análisis. De ser necesario pasar la muestra por un filtro de 0.45 µm para eliminar partículas o pase la muestra por un cartucho de SPE-IC para eliminar la interferencia de aniones como fluoruros y cloruros. Si la muestra es agua embotellada, adicionar de solución de EDA y surrogado a 1 mL de muestra previo al análisis cromatográfico. Para realizar al análisis, debe operar el equipo como se indica en el instructivo de operación correspondiente y permitir que se estabilice por lo menos 60 minutos antes de iniciar el análisis con las condiciones instrumentales establecidas. Elaborar la secuencia de análisis de acuerdo a lo indicado en la composición del lote analítico. Iniciar el análisis. Verificar que se cumpla con el control de calidad y criterios de aceptación y rechazo. La identificación cualitativa de cada compuesto determinado por éste método está basada en la comparación del tiempo de retención. Los compuestos son identificados como presentes cuando el tiempo de retención del compuesto detectado es ±5% del tiempo estándar de referencia. Si se tiene duda en la identificación por interferencias en la muestra realizar una adición de los analitos de interés a la muestra para evaluar si se mantiene la concentración y la recuperación de la adición. B.6.7.3 Análisis de datos y cálculos Una vez que se han identificado los compuestos por su tiempo de retención, efectuar la cuantificación integrando cada pico para obtener el área correspondiente. El software del instrumento genera un reporte de cuantificación (raw data) con respecto a la curva de calibración, dicho reporte contiene las áreas, los tiempos de retención y la concentración obtenida. Calcular la concentración final del compuesto detectado con la siguiente ecuación: 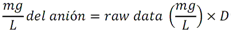 En donde: D dilución del extracto, volumen aforo (µL)/volumen tomado de la muestra (µL) B.6.7.4 Informe de prueba. Reportar el resultado como mg/L de bromato, mg/L de clorato o mg/L de clorito B.6.8 Control de calidad, calibración e interferencias B.6.8.1 Control de calidad. B.6.8.1.1 Verificación del equipo. Verificar que el equipo opere adecuadamente de acuerdo con el instructivo de operación correspondiente. Verificar las condiciones del detector: intervalo: 1 µs; temperatura de celda 35°C; temperatura de columna: 45°C; polaridad: positiva; control: local. B.6.8.1.2 Verificación de la calibración inicial. Analizar un punto medio de la curva y verificar que los valores obtenidos de la concentración para cada compuesto tengan ±25% de variación con respecto a los obtenidos en la curva de calibración inicial, de no ser así, debe preparar y analizar una nueva curva de calibración. B.6.8.1.3 Verificación de contaminación de reactivos. El blanco electrónico no debe presentar ningún tipo de contaminante que interfiera con los analitos medidos, por lo que es importante que los reactivos utilizados cumplan con las especificaciones mencionadas. Los resultados de los blancos de reactivos deben ser menores al límite de detección del método y no presentar compuestos que pudieran interferir con la identificación y/o cuantificación de los analitos medidos. Si los resultados de los blancos no cumplen con los criterios mencionados se debe localizar la fuente de contaminación y/o problema instrumental para evaluar si es necesario analizar nuevamente las muestras. Utilizar también blancos para verificar la contaminación por arrastre de muestras con altas concentraciones en la secuencia analítica. B.6.8.1.4 Verificación del proceso analítico. La muestra de control de calidad (MCC) preparada por el área de control de calidad se procesa de igual manera que las muestras. Los resultados obtenidos de la muestra control se evalúan contra los criterios de aceptación. B.6.8.1.5 Verificación de interferencias de matriz La verificación de interferencias de matriz se lleva a cabo mediante las muestras adicionadas (MA) y si lo considera necesario muestras adicionadas duplicadas MAD. Para determinar el efecto de la matriz en la recuperación del analito, calcular el porcentaje de recuperación. El % de recuperación debe estar dentro de los criterios de aceptación del 75 a 125% de exactitud y 10% DSR de precisión. Si los valores no cumplen con lo especificado, evaluar las causas, documentar las incidencias y realizar las acciones correctivas que se requieran. Para determinar el efecto de la matriz en la precisión del analito a través de la matriz adicionada duplicada (MAD), calcular la desviación estándar relativa (DSR) entre la MA y la MAD. La DPR entre la MA y la MAD debe estar dentro de los criterios de aceptación del 75 a 125% de exactitud y 10% DSR de precisión. Si los valores no cumplen con lo especificado, evaluar las causas, documentar las incidencias y realizar las acciones correctivas que se requieran. Para determinar el efecto de la matriz en la precisión del analito a través de la precisión de la matriz (M) y su matriz duplicada (MDUP), calcular la desviación estándar relativa (DSR) entre la M y la MDUP. La DPR entre la M y la MDUP debe estar dentro de los criterios de aceptación del 75 a 125% de exactitud y 10% DSR de precisión. Si los valores no cumplen con lo especificado, evaluar las causas, documentar las incidencias y realizar las acciones correctivas que se requieran. B.6.8.1.6 Verificación de la estabilidad de la curva de calibración. Verificar que la curva de calibración sea estable durante el lote analítico analizando una mezcla de verificación cada 10 corridas y observando que cumpla con los criterios establecidos. B.6.8.1.7 Composición del lote analítico El lote analítico debe estar compuesto por las siguientes muestras: B.6.8.1.7.1 blanco electrónico B.6.8.1.7.2 blanco de agua B.6.8.1.7.3 muestra de verificación de la calibración B.6.8.1.7.4 muestra de control de calidad B.6.8.1.7.5 muestra real no. 1 B.6.8.1.7.6 muestra real adicionada B.6.8.1.7.7 muestra real no. 2 B.6.8.1.7.8 muestra real duplicada B.6.8.1.7.9 muestra real no. 2 a 10 Dependiendo de los requerimientos del programa específico de control de calidad, pueden ser requeridas muestras duplicadas de campo para evaluar la precisión y exactitud del muestreo, así como las técnicas de transportación y almacenamiento de la muestra. B.6.8.1.8 La Tabla B.6-1, de este Apéndice, muestra las especificaciones de aceptación y rechazo de las verificaciones de control de calidad del método. Tabla B.6-1. Especificaciones de aceptación y rechazo de las verificaciones de control de calidad del método.
B.6.8.2 Calibración Debe llevar a cabo la estabilización del equipo a las condiciones instrumentales del método. Colocar el frasco lleno con el amortiguador de bicarbonato de sodio 4 miliMolar en la posición A. Desgasificar con helio la fase móvil por 10 minutos. Verificar que esté instalada la columna. Operar el equipo como se indica en el instructivo de operación correspondiente. Preparar la curva de calibración a partir de las disoluciones concentradas preparadas por control de calidad, se recomienda preparar dos soluciones de trabajo de acuerdo al intervalo de trabajo deseado: A partir de las disoluciones concentradas de aniones calcular la masa que será utilizada para preparar los niveles de la curva de calibración en una masa de aforo de 1 g, de acuerdo a la siguiente ecuación: 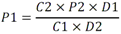 En donde: P1 peso de la disolución de referencia (g) C2 concentración del nivel de concentración requerido (mg/L) C1 concentración de la solución de referencia (mg/L) P2 peso de aforo del nivel de concentración requerido (g); será igual a 1 a menos que el volumen de aforo sea diferente a 1 g D1 densidad de la disolución de referencia (g/mL) D2 densidad del disolvente de aforo (g/mL) Calcular la concentración real de cada uno de los niveles de concentración utilizando la ecuación siguiente: 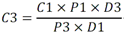 En donde: C3 concentración real de cada nivel (mg/L) C1 concentración de la mezcla de referencia utilizada (mg/L) P1 peso de la mezcla de referencia utilizada (g) P3 peso de aforo del nivel de concentración requerido (g); será igual a 1 a menos que el volumen de aforo sea diferente a 1 g D1 densidad de la mezcla de referencia (g/mL) D3 densidad del disolvente de aforo (g/mL) La Tabla B.6-2, de este Apéndice muestra la preparación de la curva de calibración
Analizar los estándares de calibración de acuerdo a las condiciones instrumentales establecidas durante la validación del método. Con los datos de concentración en mg/L y la señal obtenida en cada nivel (área), realizar una curva de concentración. Calcular las relaciones de concentración y área para cada compuesto y cada nivel, por medio de una curva de calibración calculada por mínimos cuadrados de acuerdo a la ecuación:  En donde: x concentración m factor de calibración (pendiente) y área del compuesto b ordenada al origen (expresado como respuesta) Evaluar la linealidad de la curva de calibración por medio del coeficiente de correlación, el cual debe ser mayor a 0.995. Evaluar también el coeficiente de variación del factor de calibración, el cual debe ser menor o igual al 15%. Si el coeficiente de correlación es menor a 0.995 o el coeficiente de variación del factor de calibración es mayor de 15% verificar las condiciones de operación, áreas y concentraciones para evaluar si hay algún error y corregir. Si fuera necesario, analizar nuevamente el compuesto en el o los niveles de calibración que fueran necesarios. Si detecta que el problema es la disolución estándar a partir de la cual fue preparado, reemplácelo y analice nuevamente. Una vez que ha cumplido con lo descrito anteriormente, revisar que la calibración inicial siga vigente; esto lo debe evaluar con la verificación de la estabilidad de la curva de calibración a través de la disolución estándar de un punto de la curva. De la misma manera la verificación de la estabilidad de la curva debe llevarse al inicio de cada lote analítico (posterior a la verificación del equipo y verificación de los reactivos de la fase móvil) y después de 10 horas de análisis (aproximadamente cada 10 muestras). B.6.8.3 Interferencias La necesidad de cuantificar niveles bajos de productos para la desinfección y sus precursores en presencia de niveles mucho más altos de los aniones comunes representa un problema analítico. Cualquier material iónico que coeluye con un analito de interés interferirá con la determinación de este analito. Se ha demostrado que el bromato es sujeto a interferencias positivas en algunas matrices. La interferencia es usualmente detectable como un pico aplanado. Esto puede ser a menudo eliminado al pasar la muestra a través de un cartucho de Hidrógeno (Dionex PN 039596). Este problema puede ser abordado mediante la selección de diferentes combinaciones de columna/eluyente o por la dilución del eluyente, lo cual incrementará los tiempos de retención y dispersará el cromatograma. Adicionalmente, el cloruro o cualquier otro analito no blanco presente en una concentración inusualmente alta puede sobrelaparse con el analito de interés lo suficiente para causar problemas en la cuantificación o puede ocasionar desviaciones en los tiempos de retención. La dilución de una muestra puede resolver este problema. Debe de tenerse el suficiente cuidado para evitar la acumulación de una muestra de alta concentración a la muestra subsecuente. Las interferencias del método también pueden causar la contaminación de reactivos, agua grado reactivo, cristalería de laboratorio, jeringas y otros equipos utilizados en el procesamiento de la muestra. En el análisis de muestras con alta concentración de otros aniones (fluoruros, cloruros, nitratos, etc.) o con alta fuerza iónica, algunos analitos pueden coeluir con las interferencias de la matriz, eluir sobre los picos interferentes o sufrir desplazamiento del tiempo de retención debido a la sobrecarga de los sitios activos de la columna, por lo que dependiendo de las interferencias pueden ser necesarias la confirmación por el uso de un detector alterno, columna y/o fase móvil alternas, dilución o pretratamiento de la muestras. Pueden presentarse interferencias de matriz, entre las cuales se encuentran la coelución directa de las interferencias con los analitos, la coelución con los analitos debido a altas concentraciones de las interferencias y el desplazamiento de los tiempos de retención de los analitos debida a la fuerza iónica de la muestra. Por otra parte, pueden también presentarse interferencias instrumentales. Los solventes, reactivos, material de vidrio y cualquier otro material utilizado durante el procesamiento de la muestra, pueden producir una elevación de la línea base y causar errores en la interpretación de los cromatogramas. Debe demostrarse que todos estos materiales están libres de interferencias, llevando a cabo el análisis de blancos electrónicos y de reactivos bajo las condiciones del análisis. El solvente utilizado en el equipo para el análisis (bicarbonato de sodio 4 mM) debe ser desgasificado para eliminar el aire de éste y evitar que interfiera incrementando el ruido de la línea base y disminuyendo la sensibilidad. El método utiliza el detector conductimétrico con supresor, sin embargo, algunas muestras presentan compuestos que tienen tiempos de retención tardíos por lo que pueden coeluir con los analitos determinados en la siguiente muestra y puede ser necesario realizar la confirmación de la presencia de los analitos reanalizando la muestra después de un blanco electrónico. B.6.9 Seguridad y manejo de residuos Cada reactivo utilizado en estos procedimientos debe ser tratado como un riesgo potencial para la salud y la exposición a estos materiales debe ser minimizada. Cada laboratorio es responsable de mantener un conocimiento de las regulaciones respecto a la manipulación segura de cualquier producto químico usado en este método. Debe ponerse a disposición de todo el personal involucrado en el análisis las hojas de datos de seguridad de los productos químicos. Debe de utilizarse equipo de protección personal apropiado para el manejo de muestras y estándares. Los estándares primarios deberán prepararse en una campana. Cuando el analista maneje altas concentraciones de compuestos tóxicos deberá utilizarse un respirador de gases tóxicos. Algunos de los analitos y sustancias manejados en este método pueden ser clasificados como conocidos o sospechosos carcinógenos para humanos o mamíferos por lo que cual deberán de tomarse las precauciones necesarias. B.6.10 Referencias EPA. 1997. Method 300.1 determination of inorganic anions in drinking water by ion chromatography Rev. 1.0 Standard methods for examination of water and wastewater. 4110 D. Ion Chromatographic Determination of Oxyhalides and Bromide. B.7 MÉTODOS DE PRUEBA PARA LA DETERMINACIÓN DE NITRÓGENO AMONIACAL EN AGUA PARA USO Y CONSUMO HUMANO Para la determinación de nitrógeno amoniacal (NH3-N) para el cumplimiento de esta Norma, se podrá utilizar indistintamente el: · método de prueba por titulación para la determinación de nitrógeno amoniacal en agua para uso y consumo humano (contenido en el punto B.7.1, de este Apéndice), · método de prueba para la determinación de nitrógeno amoniacal en agua para uso y consumo humano por electrodo selectivo de amoníaco (contenido en el punto B.7.2, de este Apéndice), · método de prueba para la determinación de nitrógeno amoniacal en agua para uso y consumo humano con fenato (contenido en el punto B.7.3, de este Apéndice), o · método de prueba para la determinación de nitrógeno amoniacal en agua para uso y consumo humano por colorimetría automatizada (contenido en el punto B.7.4, de este Apéndice). Los dos principales factores que determinan la selección del método para determinar amoníaco son la concentración y la presencia de interferencias. De manera general, una determinación manual directa de bajas concentraciones de amoníaco está restringida a aguas de uso y consumo humano, aguas superficiales y subterráneas de buena calidad. Por otras fuentes, y donde existen interferencias presentes o se requiere una mayor precisión, puede requerirse una destilación preliminar (contenido en el punto B.7.5, de este Apéndice). El método para la determinación de amoníaco en agua de uso y consumo humano, ya sea por titulación (punto B.7.1, de este Apéndice), por electrodo selectivo de amoníaco (punto B.7.2, de este Apéndice), con fenato (punto B.7.3, de este Apéndice) o por colorimetría automatizada (punto B.7.4, de este Apéndice), debe seleccionarse considerando la concentración de amoníaco presente en la muestra: · El procedimiento de destilación y de determinación por titulación es utilizado especialmente para concentraciones de NH3-N mayores a 5mg/L, utilizando ácido bórico como absorbente después de la destilación si el destilado será titulado. · El método por electrodo selectivo de amoníaco es aplicable en un intervalo de 0.03 a 1,400 mg de NH3-N/L. · El método manual con fenato es aplicable tanto para agua dulce como salada y es lineal a 0.6 mg NH3-N/L. Destilar en ácido sulfúrico (H2SO4) como absorbente para el método con fenato cuando se encuentran interferencias presentes. · El método automatizado es aplicable para un intervalo de 0.02 a 2 mg NH3-N/L. Asimismo, en el caso de la presencia de interferencias puede llevarse a cabo una destilación preliminar (punto B.7.5, de este Apéndice). La destilación preliminar puede llevarse a cabo cuando el método por titulación o el método con fenato son utilizados. El amoníaco destilado puede entonces ser determinado tanto colorimétricamente por el método con fenato o por titulación con una mezcla de indicadores o un medidor de pH. La selección entre el método colorimétrico y los métodos de acidimetría depende de la concentración de amoníaco. B.7.1 MÉTODO DE PRUEBA POR TITULACIÓN PARA LA DETERMINACIÓN DE NITRÓGENO AMONIACAL EN AGUA PARA USO Y CONSUMO HUMANO B.7.1.1 Principio La muestra es amortiguada en un pH de 9.5 con una disolución amortiguadora de borato para disminuir la hidrólisis de cianatos y compuestos orgánicos nitrogenados. Esta es tratada a través de una destilación preliminar (punto B.7.5, de este Apéndice) dentro de una disolución de ácido bórico. El amoníaco destilado es determinado por titulación con un estándar de H2SO4 y un indicador mixto o un medidor de pH. B.7.1.2 Alcance y aplicación El método por titulación es utilizado para la determinación de nitrógeno amoniacal en muestras de agua para uso y consumo humano que han sido previamente sometidas a un proceso de destilación preliminar (punto B.7.5, de este Apéndice). El procedimiento de destilación y de determinación por titulación es utilizado especialmente para concentraciones de NH3-N mayores a 5 mg/L, utilizando ácido bórico como absorbente después de la destilación. B.7.1.3 Equipos y materiales Cristalería. Básica de laboratorio Dispositivo para destilación (punto B.7.5, de este Apéndice) B.7.1.4 Reactivos y soluciones Agua grado reactivo. Debe utilizarse agua libre de amoníaco en la preparación de todos los reactivos y soluciones. Mezcla de indicadores. Disolver 200 mg de indicador rojo metil en 100 mL de alcohol etílico o isopropílico al 95%. Disolver 100 mg de azul de metileno en 50 mL de alcohol etílico o isopropílico al 95%. Combinar las soluciones. Esta disolución debe de ser preparada mensualmente. Disolución indicadora de ácido bórico (H3BO3). Disolver 20 g de H3BO3 en agua, añadir 10 mL de la mezcla de indicadores y diluir a 1 L. Esta disolución debe de ser preparada mensualmente. Titulante de ácido sulfúrico estandarizado 0.02N. Preparar y estandarizar la disolución. Para mayor precisión, estandarizar el titulante contra una cantidad de carbonato de sodio (Na2CO3) que ha sido incorporada a la disolución indicadora de ácido bórico para producir las condiciones reales de titulación de la muestra; 1 mL=14 X normalidad X 1 000 µg N (Para 0.02 N, 1 mL= 280 µg N). B.7.1.5 Procedimiento B.7.1.5.1 Preparación de las muestras Para seleccionar el volumen de la muestra para el método de destilación titulación puede utilizar la Tabla B.7.1-1, de este Apéndice:
B.7.1.5.2 Procedimiento analítico Llevar a cabo la destilación (punto B.7.5, de este Apéndice) utilizando una disolución indicadora de ácido bórico como absorbente para el destilado. Titular el amoniaco en el destilado con un titulante estándar 0.02N H2SO4 hasta que el indicador vire a un lavanda pálido. Llevar un blanco a través de todos los pasos del procedimiento y aplicar la corrección necesaria a los resultados. B.7.1.5.3 Análisis de datos y cálculos Calcular el nitrógeno amoniacal presente en las muestras utilizando la siguiente fórmula:  En donde: A volumen de H2SO4 titulado en la muestra (mL) B volumen de H2SO4 titulado en el blanco (mL) B.7.1.5.4 Informe de prueba. Reportar el resultado como nitrógeno amoniacal en mg/L B.7.1.6 Control de calidad, calibración e interferencias Tres muestras sintéticas que contenían amoniaco y otros constituyentes disueltos en agua reactivo fueron destiladas y analizadas por titulación, obteniendo la desviación estándar relativa y los errores relativos mostrados en la Tabla B.7.1-2
B.7.1.7 Referencias Standard methods for examination of water and wastewater. 4500-NH3 C. Titrimetric Method B.7.2 MÉTODO DE PRUEBA PARA LA DETERMINACIÓN DE NITRÓGENO AMONIACAL EN AGUA PARA USO Y CONSUMO HUMANO POR ELECTRODO SELECTIVO DE AMONÍACO B.7.2.1 Principio El método para la determinación de nitrógeno amoniacal en agua para uso y consumo humano por electrodo selectivo de amoníaco utiliza una membrana permeable a gas hidrofóbico para separar la muestra de la disolución interna del electrodo de cloruro de amonio. El amoníaco disuelto (NH3(aq) y NH4+) es convertido a NH3(aq) aumentando el pH a cerca de 11 con una base fuerte. El NH3(aq) es difundido a través de la membrana y cambia el pH de la disolución interna que es determinado por un electrodo de pH. El nivel fijo de cloruro en la disolución interna es determinado por un electrodo de ion selectivo a cloruro que sirve como un electrodo de referencia. La determinación potenciométrica es realizada con un medidor de pH que cuenta con una escala expandida de milivolts o con un medidor de ion específico. B.7.2.2 Alcance y aplicación Este método es aplicable para la determinación de NH3-N/L en un intervalo de 0.03 a 1 400 mg/L en agua potable. La medición puede ser afectada por concentraciones altas de iones disueltos, pero no por el color o la turbiedad. Por lo cual, no es necesario llevar a cabo la destilación de la muestra. Las soluciones estándar y las muestras deben tener la misma temperatura y contener cerca de los mismos niveles totales y especies disueltas. El método por electrodo selectivo de amoníaco responde lentamente debajo de 1 mg NH3-N/L, por lo que deben utilizarse tiempos largos de inmersión del electrodo (2 o 3 minutos) para obtener lecturas estables. B.7.2.3 Equipos y materiales Agitador magnético. Aislado térmicamente con barra de agitación cubierta de tetrafluoroetileno (TFE) Electrodo selectivo de amoníaco Potenciómetro. Medidor de pH con una escala expandida en milivoltios (mV) con una resolución de 0.1 mV entre -700 mV y +700 mV o un medidor de ion específico. B.7.2.4 Reactivos y soluciones Agua reactivo libre de amoníaco Hidróxido de sodio 10N Soluciones estándar de cloruro de amonio. Disolución NaOH/EDTA, 10N. Disolver 400 g de NaOH en 800 mL de agua. Añadir 45.2 g de la sal tetrasódica del ácido etilendiaminotetracético, tetrahidratada (Na4EDTA 4H2O) y agitar hasta disolver. Enfriar y diluir a 1 000 mL Disolución concentrada de cloruro de amonio (NH4Cl). Disolver 3.819 g de cloruro de amonio (NH4Cl) anhidro (secado a 100°C) en agua y diluir a 1 000 mL; 1 mL=1 mg N=1.22 mg de amoniaco. B.7.2.5 Procedimiento B.7.2.5.1 Preparación de estándares Preparar una serie de disoluciones estándar por diluciones decimales a partir de una disolución patrón concentrada de NH4Cl que cubran las concentraciones de 1000, 100, 10, 1 y 0.1 mg NH3-N/L. B.7.2.5.2 Calibración de potenciómetro Poner 100 mL de cada disolución estándar en un vaso de precipitados de 150 mL. Sumergir el electrodo en el estándar de concentración más baja y mezclar con un agitador magnético. Limitar la velocidad del agitador para minimizar la posible pérdida de amoníaco de la disolución. Mantener ella misma velocidad de agitación y una temperatura de 25 °C durante la calibración y determinación. Añadir un volumen suficiente de disolución NaOH 10N (usualmente 1 mL es suficiente) para aumentar el pH a cerca de 11. Si la presencia de plata o mercurio es posible, usar una disolución de NaOH/EDTA en lugar de disolución NaOH. Si es necesario añadir más de 1 mL de disolución NaOH o NaOH/EDTA registrar el volumen utilizado, debido a que es requerido en los cálculos. Mantener el electrodo en la disolución hasta obtener una lectura estable en milivolts. No añadir disolución NaOH antes de sumergir el electrodo, debido a que el amoniaco puede perderse a partir de la disolución básica. Repetir el procedimiento con el resto de los estándares, procediendo de la concentración más baja a la más alta. Esperar hasta que la lectura se estabilice (al menos 2 o 3 minutos) antes de registrar los milivolts para los estándares y las muestras que contienen una concentración 1 mg NH3-N/L. B.7.2.5.3 Preparación de la curva estándar Obtener la gráfica de la ecuación de concentración con el log de la concentración del de NH3-N/L en mg en el eje x y el potencial en milivolts en el eje y. Si el electrodo funciona apropiadamente un cambio de 10 veces la concentración de NH3-N produce un cambio en el potencial de cerca de 59 mV. B.7.2.5.4 Procedimiento analítico Diluir si es necesario para llevar la concentración de NH3-N dentro del intervalo de la curva de calibración. Colocar 100 mL de muestra en un vaso de precipitados de 150 mL y seguir el mismo procedimiento descrito para la calibración del electrómetro. Registrar el volumen de disolución NaOH 10 N añadido. Leer la concentración de NH3-N a partir de la curva estándar. B.7.2.5.5 Análisis de datos y cálculos Calcular el nitrógeno amoniacal presente en las muestras utilizando la siguiente fórmula: 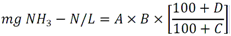 En donde: A factor de dilución B concentración de NH3-N/L, mg/L, a partir de la curva de calibración C volumen de disolución NaOH 10N añadida a los estándares de calibración, mL D volumen de disolución NaOH 10N añadida a la muestra, mL B.7.2.5.6 Informe de prueba. Reportar el resultado como nitrógeno amoniacal en mg/L B.7.2.6 Referencias Standard methods for examination of water and wastewater. 4500-NH3 D. Ammonia-selective electrode method. B.7.3 MÉTODO DE PRUEBA PARA LA DETERMINACIÓN DE NITRÓGENO AMONIACAL EN AGUA PARA USO Y CONSUMO HUMANO CON FENATO B.7.3.1 Principio Un compuesto de color azul intenso es formado por la reacción de amoniaco, hipoclorito y fenol, catalizada por una sal de manganeso. El ion amonio reacciona con el hipoclorito y fenol en medio alcalino en presencia de nitroferricianuro para producir un compuesto colorido azul de indofenol. B.7.3.2 Alcance y aplicación El método con fenato es utilizado para la determinación de nitrógeno amoniacal en muestras de agua para uso y consumo humano que han sido previamente sometidas a un proceso de destilación preliminar (contenido en el punto B.7.5 de este Apéndice) para eliminar interferencias. Destilar en ácido sulfúrico (H2SO4) como absorbente para el método con fenato cuando se encuentran interferencias presentes. El método manual con fenato es aplicable tanto para agua dulce como salada y es lineal a 0.6mg NH3-N/L. B.7.3.3 Equipos y materiales Agitador magnético Espectrofotómetro, para usar a 630 nm con un paso de luz de 1 cm o mayor. B.7.3.4 Reactivos y soluciones Agua libre de amoníaco. Utilizar para la preparación de todos los reactivos. Reactivo de ácido hipocloroso. A 40 mL de agua reactivo añadir 10 mL de una disolución al 5-6% de hipoclorito de sodio (NaOCl) preparada o adquirida comercialmente. Ajustar el pH a 6.5-7.0 con HCl. Preparar este reactivo inestable semanalmente. Reactivo de fenato. Disolver 2.5 g de NaOH y 10g de fenol C6H5OH en 100 mL de agua reactivo. Debido a que este reactivo se oscurece preparar semanalmente. Debe tenerse precaución al utilizar el fenol. Disolución patrón concentrada de amonio. Disolver 381.9 mg de NH4CL anhidro secado a 100 °C, en agua reactivo y diluir a 1 000 mL; 1 mL=100 µg N=122 µg NH3 Solución patrón de trabajo de amonio. Diluir 5 mL de la solución concentrada de amonio en 1 000 mL de agua reactivo; 1 mL= 0.500 µg N=0.610 µg NH3 Disolución de sulfato manganoso (MnSO4·H2O) 0.003 M. Disolver 50 mg de sulfato manganoso (MnSO4·H2O) en 100 mL de agua B.7.3.5 Procedimiento B.7.3.5.1 Procedimiento analítico A una muestra de 10 mL contenida en un vaso de precipitados de 50 mL añadir una gota (0.05mL) de disolución MnSO4. Colocar sobre un agitador magnético y añadir 0.5 mL de disolución de ácido hipocloroso. Inmediatamente añadir por goteo 0.6 mL de reactivo de fenato. Añadir el reactivo sin tardarse utilizando una pipeta beral para una conveniente adición del reactivo. Marcar la pipeta para ácido hipocloroso al nivel de 0.5 mL y liberar el reactivo de fenato de la pipeta que ha sido calibrada contando el número de gotas previamente contadas que son equivalentes a un total de 0.6 mL. Agitar vigorosamente durante la adición de los reactivos. Debido a que la intensidad del color es afectado por el tiempo que llevan preparados los reactivos, llevar un blanco y un estándar a través del procedimiento con cada lote de muestras. Medir la absorbancia utilizando un blanco de reactivos para establecer la lectura cero en el espectrofotómetro. La formación de color se completa en 10 minutos y es estable por al menos 24 horas. A pesar de que el color azul tiene un máximo de absorbancia a 630 nm, pueden realizarse determinaciones satisfactorias en la región de 600 a 660 nm. Preparar una curva de calibración en el intervalo de 0.1 a 5 µg de NH3-N, tratado los estándares de la misma manera que las muestras. B.7.3.5.2 Análisis de datos y cálculos Graficar las lecturas de absorbancia contra concentración, obtener la ecuación de la recta, y comparar las lecturas de las muestras con esta. La concentración de amoníaco puede ser calculada utilizando la siguiente ecuación:  En donde: A absorbancia de la muestra B NH3-N en el estándar, µg C absorbancia del estándar S volumen de la muestra utilizado, mL D volumen del destilado total colectado, mL, incluyendo el ácido absorbente, el agente neutralizante y al agua libre de amoniaco añadida E volumen del destilado utilizado para el desarrollo de color, mL El radio D/E aplica sólo para muestras destiladas B.7.3.5.3 Informe de prueba Reportar el resultado como nitrógeno amoniacal en mg/L B.7.3.6 Interferencias Una alcalinidad mayor a 500 mg como carbonato de calcio (CaCO3)/L, una acidez encima de 100 mg como CaCO3/L y la turbidez interfieren con el método, Por lo cual es necesario remover estas interferencias con una destilación preliminar (ver punto B.7.5, de este Apéndice). B.7.3.7 Referencias Standard methods for examination of water and wastewater. 1975. 4500-NH3 D. Phenate Method. 14 ed. B.7.4 MÉTODO DE PRUEBA PARA LA DETERMINACIÓN DE NITRÓGENO AMONIACAL EN AGUA PARA USO Y CONSUMO HUMANO POR COLORIMETRÍA AUTOMATIZADA B.7.4.1 Definiciones y términos Análisis de blanco analítico, someter una alícuota de agua reactivo a todo el proceso de análisis por el cual pasa una muestra real. Los laboratorios deben realizar los análisis de blancos para corregir la señal de fondo del sistema de medición. El análisis de blancos se realizará en forma periódica o con cada lote de muestras según lo requiera el método. Blanco, al agua reactivo o matriz equivalente a la que no se le aplica ninguna parte del procedimiento analítico y sirve para evaluar la señal de fondo. Blanco analítico o de reactivos, al agua reactivo o matriz equivalente que no contiene, por adición deliberada, la presencia de ningún analito o sustancia por determinar, pero que contiene los mismos disolventes, reactivos y se somete al mismo procedimiento analítico que la muestra problema. Blanco fortificado, al estándar preparado a partir de una muestra blanco (agua o agua reactivo) a la cual se agrega el analito en una concentración conocida y no necesariamente de una fuente diferente a la que se usa para la calibración. Se prepara al menos uno por cada lote de muestras. Calibración, al conjunto de operaciones que establecen, bajo condiciones específicas, la relación entre los valores de una magnitud indicados por un instrumento o sistema de medición, o los valores representados por una medida materializada y los valores correspondientes de la magnitud, realizados por los patrones, efectuando una corrección del instrumento de medición para llevarlo a las condiciones iniciales de funcionamiento. Desviación estándar experimental, para una serie de mediciones del mismo mensurando, es la magnitud que caracteriza la dispersión de los resultados. Disolución estándar, a la disolución de concentración conocida preparada a partir de un patrón primario. Disolución madre, corresponde a la disolución de máxima concentración en un análisis. Es a partir de esta disolución que se preparan las disoluciones de trabajo. Disolución patrón, a la disolución de concentración conocida preparada a partir de un patrón primario. Exactitud, proximidad de concordancia entre el resultado de una medición y un valor verdadero del mesurado. Límite de detección del método (LDM), a la concentración mínima del analito que puede detectarse pero no necesariamente cuantificarse bajo las condiciones de operación establecidas. Este límite de detección generalmente se logra por analistas experimentados con equipo bien calibrado y bajo condiciones no rutinarias. Límite práctico de cuantificación (LPC), a la concentración mínima del analito que puede determinarse con un nivel de confianza predeterminado en condiciones rutinarias de operación. Este límite puede establecerse entre 5 a 10 veces el LDM. Material de referencia, al material o sustancia en el cual uno o más valores de sus propiedades son suficientemente homogéneos y bien definidos, para ser utilizadas para la calibración de aparatos, la evaluación de un método de medición o para asignar valores a los materiales. Material de referencia certificado, al material de referencia, acompañado de un certificado, en el cual uno o más valores de las propiedades están certificados por un procedimiento que establece la trazabilidad a una realización exacta de la unidad en la cual se expresa los valores de la propiedad, y en el que cada valor certificado se acompaña de una incertidumbre con un nivel declarado de confianza. Medición, al conjunto de operaciones que tiene por objeto determinar el valor de la magnitud. Mensurando, a la magnitud particular sujeta a medición. Muestra de control de calidad (MCC), a la muestra que contiene todos o un subgrupo de los analitos del método a concentraciones conocidas. Las MCC se obtienen de una fuente externa al laboratorio o bien se pueden preparar por el analista de una fuente de estándares diferentes de la fuente de los estándares de calibración. Parámetro, a la variable que se utiliza como referencia para determinar la calidad del agua. Patrón de medición, al material de referencia, instrumento de medición, medida materializada o sistema de medición destinado a definir, realizar, conservar o reproducir una unidad o uno o más valores de una magnitud para utilizarse como referencia. Patrón de referencia, al patrón en general de la más alta calidad metrológica disponible en un lugar dado, o en una organización determinada de la cual se derivan las mediciones realizadas en dicho lugar. Patrón de trabajo, al patrón que es usado rutinariamente para calibrar o controlar las medidas materializadas, instrumentos de medición o los materiales de referencia. Patrón nacional de medición, al patrón reconocido por una decisión nacional en un país, que sirve de base para asignar valores a otros patrones de la magnitud concerniente. Patrón primario, al patrón que es designado o reconocido ampliamente como un patrón que tiene las más altas cualidades metrológicas y cuyo valor es aceptado sin referencia a otros patrones de la misma magnitud. Patrón secundario, al patrón cuyo valor es establecido por comparación con un patrón primario de la misma magnitud. Precisión, al grado de concordancia entre resultados analíticos individuales cuando el procedimiento analítico se aplica repetidamente a diferentes alícuotas o porciones de una muestra homogénea. Usualmente se expresa en términos del intervalo de confianza o incertidumbre. B.7.4.2 Símbolos y términos abreviados CCC verificación de calibración continúa ICV verificación inicial de la calibración MCC muestra de control de calidad LDM límite de detección del método LPC límite práctico de cuantificación µuA micro unidad de absorbancia botón de inicio de línea base botón de inicio de análisis B.7.4.3 Principio El amonio reacciona con el hipoclorito para generar cloramina. Ésta reacciona con el fenol en medio alcalino y en presencia de nitroferricianuro para producir azul de indofenol, cuya concentración es proporcional a la concentración de amonio en la muestra (Fig. B.7.4-1, de este Apéndice). La concentración del azul de indofenol se cuantifica leyendo a una longitud de onda de 660 nm. 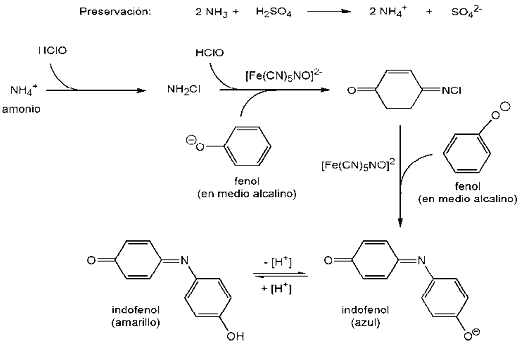 Fig. B.7.4-1. Formación del indofenol de color azul producto de la reacción del ión amonio con hipoclorito y fenol en medio básico en presencia de nitroferricianuro. B.7.4.4 Alcance y aplicación Este método se usa para determinar la concentración de nitrógeno amoniacal (N-NH3) en un intervalo de 0.01 a 2.0 mg/L en agua potable, natural, subterránea y superficial. El intervalo de trabajo se puede extender hacia concentraciones más altas mediante la dilución de las muestras. B.7.4.5 Equipos y materiales Sólo se mencionan los equipos y materiales que no son de uso común en el laboratorio analítico. Analizador automático, que incluya bomba peristáltica de canales múltiples, automuestreador, detector fotométrico con celda de flujo, sistema de recopilación de datos y cartucho configurado para el análisis de nitrógeno amoniacal (Fig. B.7.4-2, de este Apéndice). Si se usa el equipo en la modalidad de FIA, el equipo debe contar con la válvula apropiada. El siguiente diagrama considera el uso del equipo automatizado en su modalidad de flujo segmentado (SFA).  Fig. B.7.4-2. Configuración del cartucho para el análisis de N-NH3 Agitador rotatorio Balanza Embudos de filtración Material de vidrio lavado de acuerdo al instructivo de control de calidad. Potenciómetro o tiras reactivas indicadoras de pH Papel filtro, de tamaño de poro de 2.5 µm o menor. B.7.4.6 Reactivos y soluciones B.7.4.6.1 Los reactivos empleados en este método se indican en la Tabla B.7.4-1, de este Apéndice.
B.7.4.6.2 Preparación de reactivos Para mejores resultados, filtrar con membrana de 0.45µm todas las disoluciones de los reactivos preparados antes de usarlos. Se puede hacer uso de agua tipo I y desgasificada, para la obtención de mejores resultados. B.7.4.6.2.1 Agua reactivo. Debe ser agua libre de amonio, pH de 5.0 a 8.0 y conductividad máxima de 5 µS/cm a 25ºC. Adicionalmente se puede hacer un tratamiento de desgasificación del agua a utilizar si el analista lo considera prudente con base en el ruido instrumental obtenido en el equipo. (Se sugiere realizar con ruido instrumental > 1000 µuA). La desgasificación puede realizarse a través de someter el agua desionizada a un vacío fuerte durante 15 a 20 minutos; agitar magnéticamente o someter a un ultrasonido por 20 minutos; purgar el agua con nitrógeno gaseoso (u otro gas inerte) con ayuda de un material poroso por 5 minutos o hervir el agua desionizada en un matraz Erlenmeyer por 15 minutos y dejar enfriar a temperatura ambiente cubriendo la boca del matraz con un vaso de precipitados invertido. Una vez desgasificada el agua, guardar en un contenedor cerrado para evitar que absorba gases atmosféricos. B.7.4.6.2.2 Disolución de arranque / disolución de lavado. Esta disolución se usa para ambos propósitos. Añadir 0.5 mL de Brij-35 a 500 mL de agua desionizada. Mezcle suavemente. B.7.4.6.2.3 Reactivo concentrado complejante, Sal disódica del ácido etilendiaminotetraacético dihidratada al 5% p/v (1 L). Disolver 50 g de la Sal disódica del ácido etilendiaminotetraacético dihidratada y aproximadamente 1 g de hidróxido de sodio (aproximadamente 6 perlas) en aproximadamente 800 mL de agua desionizada. Llevar a un volumen final de 1 L con agua desionizada. B.7.4.6.2.4 Reactivo complejante de trabajo (250 mL). Tomar 250 mL del reactivo complejante de EDTA, añadir 250 µL de detergente Brij-35 y mezclar suavemente. Este volumen es suficiente para un análisis de 8 horas. Preparar esta disolución cada vez que se analice un lote. B.7.4.6.2.5 Disolución concentrada de hidróxido de sodio 10 N (1 L). Mientras agita, añadir cuidadosamente 400 g de hidróxido de sodio sólido a aproximadamente 500 mL de agua. Dejar enfriar a temperatura ambiente y diluir a 1 000 mL con agua. Mezclar bien. Guardar en un recipiente de polietileno. Es importante considerar que al disolver el hidróxido de sodio en agua se libera una gran cantidad de calor por lo que deben tomarse las precauciones necesarias. B.7.4.6.2.6 Fenol alcalino (1 L). Mientras agita, añada cuidadosamente 80 mL de la disolución de NaOH 10 N a aproximadamente 700 mL de agua. Enfríe la disolución en un baño de hielo si es necesario. Mientras agita, añada cuidadosamente 83 g de fenol en pequeñas porciones, enfriando la disolución después de cada adición. Lleve a un volumen final de 1 000 mL con agua desionizada y mezcle bien. Guarde en un frasco ámbar a 4 °C. Esta disolución es estable hasta por 6 semanas si se guarda en estas condiciones. Al disolver hidróxido de sodio o fenol en agua se libera una gran cantidad de calor. Tome las precauciones necesarias. La disolución de fenol alcalino debe presentar un color paja tenue. Deséchela si se torna de color ámbar oscuro. B.7.4.6.2.7 Disolución de hipoclorito de sodio de trabajo (100 mL). Tome 50 mL de la disolución de hipoclorito de sodio comercial (5% de cloro disponible) y mézclelos con 30 mL de agua desionizada. Llevar a un volumen final de 100 mL con agua y mezclar bien. Debido a la volatilidad del hipoclorito de sodio, no es posible almacenar esta disolución. Prepare este reactivo cada vez que realice un análisis. B.7.4.6.2.8 Nitroferricianuro de sodio (1 L). Pesar y disolver 0.5 g de nitroferricianuro de sodio dihidratado en aproximadamente 800 mL de agua. Llevar a un volumen final de 1 000 mL y mezcle bien. Guardar en un frasco ámbar. Esta disolución es estable hasta por 6 semanas si se mantiene a 4 °C. B.7.4.6.2.9 Hidróxido de sodio, disolución 10N (1L). Mientras se agita, adicionar cuidadosamente 400 g de hidróxido de sodio a aproximadamente 500 ml de agua desionizada, en un vaso de precipitados inmerso en baño refrigerante. Diluir con agua desionizada hasta alcanzar un volumen final de 1 000 ml, envasar en recipiente de plástico. B.7.4.6.2.10 Disolución madre de 1 000 mg/L de nitrógeno amoniacal (N-NH3). Preparación volumétrica. Pesar con precisión 0.471 7 g de sulfato de amonio, (NH4)2SO4, secado previamente a 105 ± 3 °C que cuente con la pureza necesaria para ser considerado como material de referencia y el certificado que lo demuestre. Disolver en aproximadamente 80 mL de agua desionizada dentro de un matraz aforado y verificado. Diluya hasta 100 mL con agua desionizada. Guarde en un frasco y preserve con 3 gotas de cloroformo. Esta disolución es estable por tres meses si se conserva en refrigeración. La preparación de esta solución madre es recomendable hacerla gravimétricamente, la estabilidad de la solución puede ser hasta de 6 meses en condiciones de almacenamiento adecuadas. B.7.4.6.2.11 Disolución madre de 1 000 mg/L de nitrógeno amoniacal (N-NH3). Preparación gravimétrica. Realizar la preparación de la disolución en un contenedor de plástico y de volumen adecuado para este fin. Agregar agua desionizada cuidadosamente hasta alcanzar un peso de 100 g. B.7.4.7 Procedimiento B.7.4.7.1 Recolección, conservación y almacenamiento de muestras Las muestras se colectan en envases de vidrio o plástico limpios. El volumen de muestra debe ser suficiente para garantizar que se tenga una muestra representativa, que permita el análisis de duplicados si es necesario y que minimice la cantidad de desechos generados. Se sugiere tomar un volumen de muestra entre 50 y 100 mL. Es posible determinar la concentración de nitrógeno amoniacal en las muestras sin preservar si se analizan inmediatamente después de su recolección. Si no es posible analizar las muestras inmediatamente preservarlas añadiendo ácido sulfúrico (H2SO4) hasta obtener un pH < 2 y mantenerlas a 4 °C hasta su análisis. Las muestras preservadas deben analizarse tan pronto como sea posible después de su recolección. Pueden almacenarse hasta por 28 días si se mantienen a 4 °C. Ajuste el pH de las muestras entre 5 y 8 antes de analizarlas con NaOH. B.7.4.7.2 Preparación de muestras Tome un volumen suficiente de muestra para filtrarlo y recuperar un volumen de filtrado de al menos 9 mL. Generalmente un volumen de 50 mL de muestra es suficiente. Si se sospecha o se sabe de la presencia de cloro residual se trata la muestra en la filtración agregando una pizca de tiosulfato de sodio, sulfito de sodio o algún otro agente equivalente. Filtrar las muestras con papel filtro de poro mediano. Un tamaño de poro de 2.5 µm es suficiente, aunque se puede usar un tamaño de poro más pequeño. Las muestras aparentemente limpias y que sean además de origen potable pueden no ser filtradas antes de neutralizarlas. Si las muestras filtradas aún presentan coloración, adicione una pizca de carbón activado, agite y fíltrelas nuevamente. Verifique el pH de las muestras con tiras reactivas indicadoras de pH o con un potenciómetro. Ajuste el pH de las muestras entre 5 y 8 con NaOH 10 N. Transfiera el líquido filtrado a viales de vidrio de 10mL para su análisis. En caso de que se tengan muestras de hielo, se toma un fragmento de éste buscando que la alícuota sea representativa y se coloca en un envase adecuado para contenerlo, después de unos minutos que la muestra ya sea líquida se envasa en tubos de 10 mL para su análisis. En caso de tener una muestra de agua envasada, se toma alícuota directamente en el recipiente original y envasando en tubos de 10 mL para su análisis posterior. B.7.4.7.3 Preparación del autoanalizador Configure el equipo como lo indica el Manual del Usuario respectivo y asegúrese de que todos los módulos estén encendidos. Verifique que las conexiones de las mangueras estén configuradas en el cartucho como muestra el diagrama de la Figura B.7.4-2. Asegúrese de tener instalado el filtro de 660 nm en el detector. Active el calentador del cartucho y verifique que la temperatura sea de 50 °C. Llene el depósito de enjuague del automuestreador con agua. B.7.4.7.4 Estabilización del equipo Abrir el programa del equipo y asegurarse que la cánula del automuestreador se introduzca en la cavidad de enjuague. Conecte la manguera del reactivo complejante de trabajo (EDTA) a un recipiente con disolución de arranque/lavado y las mangueras del resto de los reactivos a un recipiente con agua. Asegure las grapas de todas las mangueras en la bomba presionándolas hacia abajo hasta que escuche un "clic" y levante todas las palancas correspondientes para presionar las mangueras contra los rodillos de la bomba peristáltica. Accione la bomba a una velocidad de 40% permitiendo que el agua y la disolución de arranque fluyan por todo el sistema. Asegúrese de que no existan fugas en las conexiones, que no haya mangueras presionadas y que los flujos en las mangueras sean constantes. B.7.4.7.5 Verificación de la línea base Crear o cargar un método apropiado para el análisis de N-NH3 en el programa del equipo. Consultar el Manual del Usuario respectivo del programa para mayor referencia. Crear una secuencia que incluya las muestras que conforman el lote analítico y los controles de calidad. Consultar el Manual del Usuario respectivo del programa para mayor referencia. Seleccionar la opción del menú de la ventana principal del programa del equipo. Introducir el nombre y clave del analista, seleccionar el método y la secuencia a usarse en el análisis e indique un nombre y una ruta para guardar los resultados del análisis. Comenzar el monitoreo de la línea base presionando el botón respectivo que generalmente tiene el símbolo () ubicado en el margen izquierdo de la pantalla de obtención de datos. Monitorear la línea base por unos minutos. Su magnitud no debe sobrepasar las 1000 µuA y su deriva (desplazamiento hacia arriba o hacia abajo) no debe ser mayor a las 500 µuA en 300 segundos. Si observa fluctuaciones grandes en la línea base o una deriva continua probablemente se deba a la presencia de burbujas en la celda de flujo. Extraer estas burbujas tomando la manguera a la salida del flujo de la celda y forme un bucle con ella. Presionar con los dedos el punto donde se sobreponen las dos secciones de la manguera y tire ligeramente mientras presiona, sosteniendo con la otra mano la porción de la manguera más próxima a la celda de flujo. Observar si se estabiliza la línea base. Repita esta operación hasta que la línea base sea estable. Conectar todas las mangueras a los recipientes de los reactivos correspondientes y verifique nuevamente las condiciones de la línea base. B.7.4.7.6 Análisis Coloque la(s) gradilla(s) con los estándares y las muestras en el automuestreador. Verifique que las posiciones de los tubos coincidan con las especificadas en la secuencia de análisis. La matriz de los calibrantes, blancos y muestras sintéticas deben tener la misma matriz que las muestras analizadas. Inicie el análisis presionando el botón respectivo que generalmente tiene el símbolo () ubicado en el margen izquierdo de la pantalla de obtención de datos. Esto inicia el análisis de acuerdo a la secuencia cargada. De ser necesario, realice las diluciones de las muestras que se encuentren fuera del intervalo de trabajo e inclúyalas al final de la secuencia. Anexe también al final de la secuencia las muestras para las que se hayan obtenido resultados poco confiables debido a anomalías durante su análisis, tal como el arrastre de la muestra anterior o el paso de alguna burbuja por el detector. Para anexar muestras a la secuencia abra la tabla durante el análisis del lote presionando el botón correspondiente ubicado en el margen izquierdo de la pantalla de obtención de datos e introduzca la información en las posiciones disponibles al final de la secuencia. Una vez terminado el análisis, cambie la manguera del reactivo complejante de trabajo (EDTA) a un recipiente con disolución de arranque/lavado y las mangueras del resto de los reactivos a un recipiente con agua desionizada. Deje fluir por todo el sistema el agua desionizada y la disolución de lavado por 15 minutos para limpiarlo. Detenga la bomba, libere las grapas de las mangueras en la bomba peristáltica, apague el equipo y vacíe el recipiente de desechos. Si observa precipitación al conectar la línea del fenol alcalino, puede deberse a una baja calidad de los reactivos, en particular del EDTA, o demasiado Brij-35. La precipitación al conectar el fenol alcalino también puede presentarse si la muestra contiene calcio o magnesio en tal cantidad que sobrepase la capacidad complejante del EDTA. Si eso ocurre, aumente la concentración de EDTA en el reactivo complejante. Se sugiere aumentar la cantidad en un 50%, ya que demasiado EDTA ocasiona un aumento considerable en el ruido. El pH del medio cuando sale de la celda de flujo debe ser aproximadamente de 12. Verifíquelo con tiras reactivas indicadoras de pH. Para mejorar el desempeño del método a bajas concentraciones se recomienda: · Preparar una dilución 1:10 de la disolución complejante de trabajo. · Disminuir el intervalo de trabajo de manera que se acerque a la concentración de las muestras. · Aumentar el tiempo de duración del ciclo en las condiciones del método para mejorar el desempeño del método a concentraciones altas. B.7.4.7.7 Análisis de datos y cálculos La curva de calibración permite al equipo calcular automáticamente la concentración de las muestras. Las unidades de los valores arrojados son las mismas que se usaron para los estándares, que son mg/L de N-NH3. B.7.4.7.8 Informe de prueba. Reportar el resultado como mg/L de nitrógeno amoniacal (N-NH3). B.7.4.8 Control de calidad, calibración e interferencias B.7.4.8.1 Control de Calidad B.7.4.8.1.1 Verificación del equipo. Asegure el buen funcionamiento del equipo verificando los Indicadores de Calidad de la técnica. B.7.4.8.1.2 Verificación de la calibración. Si para cuantificar la concentración de analito en las muestras y los controles de calidad se emplea una curva de calibración cargada en el programa obtenida anteriormente (en la validación parcial del método), se emplea la respuesta que arroja el pico de sincronía para evaluar la veracidad de la curva. Se compara la respuesta obtenida para este pico con el promedio de las respuestas obtenidas en la validación del método para un estándar de la misma concentración. La diferencia entre estas dos respuestas no debe ser mayor al 15%. Si se hace uso de una curva de calibración nueva para cuantificar la concentración de analito en las muestras y los controles de calidad en un lote, se debe preparar un estándar de verificación inicial de la calibración (ICV), el cual contiene al analito en una concentración conocida y que se encuentra dentro del intervalo de trabajo. Se sugiere que esta concentración sea menor o igual al punto medio de la curva de calibración. Este estándar debe ser preparado a partir de una fuente alterna a la que se empleó para construir la curva de calibración. B.7.4.8.1.3 Verificación de contaminación de reactivos. Se prepara un blanco de reactivos tomando una alícuota de agua desionizada y sometiéndola al mismo procedimiento de preparación y de análisis que el resto de las muestras. La lectura para este blanco de reactivos debe arrojar un valor menor al límite de detección del método. Se debe incluir una muestra blanco por cada 20 muestras reales. B.7.4.8.1.4 Verificación del proceso analítico. Se emplean diferentes muestras para verificar el proceso analítico: · Verificación inicial de la calibración (ICV). · Verificación de calibración continua (CCC). Con el propósito de comprobar que la calibración verificada al inicio se mantenga vigente durante todo el análisis del lote, se debe analizar a intervalos regulares una disolución estándar de concentración conocida. Se sugiere que ésta sea de concentración menor o igual al punto intermedio de la curva. El recobro entre el valor nominal de esta disolución y el valor obtenido en cada medición no debe ser mayor al 10%. · Verificación de la linealidad de la curva de calibración. El programa del equipo ajusta una ecuación a los pares de datos obtenidos para las muestras con las que se construyó la curva de calibración. · Este ajuste suele ser de primer orden, aunque puede ser de segundo orden si el intervalo de trabajo es demasiado amplio. Además de la función ajustada, el programa arroja un coeficiente de correlación lineal, el cual debe ser igual o mayor a 0.995. · Muestra control de calidad. Se analiza una muestra sintética de concentración conocida (muestra QC), siendo lo más recomendable prepararla a partir de una fuente diferente a la curva de calibración por cada 20 muestras reales. El recobro entre el resultado obtenido y el valor nominal de esta muestra no debe ser mayor al 10%. · Verificación de repetibilidad de los análisis. Se selecciona al menos una muestra por cada matriz del lote analizado para prepararla y analizarla por duplicado o bien el 10% del total de muestras. Con ello se verifica la repetibilidad del proceso al cual se someten las muestras. La diferencia porcentual relativa entre los dos valores obtenidos no debe superar el 20%. · Verificación de interferencias de matriz. Se deben adicionar al menos dos muestras por cada matriz del lote analizado. Las muestras seleccionadas se adicionan con una concentración conocida de analito, la cual se recomienda que sea igual a la establecida como máximo permisible por alguna entidad reguladora o, en su caso, que sea entre una y cinco veces la concentración del analito presente originalmente en la muestra o si este dato se desconoce se recomienda adicionar una concentración del punto medio hacia abajo de la curva de calibración. Comparando las concentraciones de analito en la muestra sin adicionar y en la adicionada se obtiene un recobro de la adición, el cual debe de estar entre el 80 y el 120 %. Se recomienda que las muestras que se dupliquen sean las mismas que se adicionan. Y si se prepara un blanco fortificado, que éste tenga la misma concentración de las muestras adicionadas. · Composición del lote. El lote analítico debe conformarse como se indica en la Tabla B.7.4-2, de este Apéndice.
Dependiendo de los requerimientos del programa específico de control de calidad, pueden ser requeridas muestras duplicadas de campo para evaluar la precisión y exactitud del muestreo, así como las técnicas de transportación y almacenamiento de la muestra. La Tabla B.7.4-3, de este Apéndice resume los controles de calidad mencionados en esta sección, así como sus criterios de aceptación.
B.7.4.8.2 Calibración Si para cuantificar la concentración de N-NH3 se emplea una curva obtenida anteriormente en la validación parcial del método, solamente verifique la calibración del instrumento. Esto es preparando una disolución conocida como pico de sincronía que es equivalente a una concentración del último punto de la curva de calibración y un estándar de verificación continua de la calibración los cuales se recomienda sean de una fuente diferente con la que se validó el método. Si se requiere de una curva de calibración nueva para cuantificar la concentración de analito en las muestras y en los controles de calidad de un lote, o para realizar una nueva validación parcial del método, prepárela usando material volumétrico verificado, o bien, gravimétricamente empleando una balanza calibrada. Disolución intermedia de N-NH3. Tome 1 mL de la disolución madre de 1 000 mg/L de N-NH3 y lleve a un volumen final de 100 mL con agua desionizada. Con ello se obtiene una concentración de 10 mg/L de N-NH3. En la Tabla B.7.4-4, de este Apéndice, se muestran las cantidades sugeridas para obtener una curva de N-NH3 en el intervalo de 0.01 a 5 mg/L a partir de la disolución intermedia de N-NH3.
Prepare el equipo para el análisis e incluya en la secuencia los puntos de calibración preparados, introduciendo las concentraciones calculadas para cada punto. El equipo registra la respuesta obtenida para cada punto y la asocia a la concentración especificada por el usuario, con lo que construye y almacena de manera automática una curva de calibración. Esta curva es usada después para obtener la concentración de las muestras desconocidas. La verificación de la calibración durante el análisis del lote se realiza mediante la CCC. B.7.4.8.3 Interferencias Al tratarse de una técnica fotométrica, la turbidez en las muestras interfiere con la capacidad del detector para registrar la absorción real de la muestra. Filtre las muestras turbias antes de analizarlas. La precipitación de hidróxidos o carbonatos de calcio y magnesio debido al pH del medio de reacción interfiere con las lecturas fotométricas. Evite la formación de estos sólidos mediante el uso de ácido etilendiamintetraacético (EDTA). Las muestras que presenten por su naturaleza una absorbancia a 660 nm presentarán una interferencia positiva. La intensidad del color generado en las reacciones que se muestran en la figura 1 es sensible al pH. Ajuste el pH de todas las muestras entre 5 y 7 antes de analizarlas. Los cianatos pueden ser encontrados en ciertos efluentes industriales, estos pueden hidrolizarse en cierta medida incluso a pH 9.5 por lo que se puede realizar una destilación para eliminar esta interferencia. El cloro residual debe ser removido mediante un pretratamiento de la muestra con tiosulfato de sodio, sulfito de sodio o bien otro reactivo decloronizador. B.7.4.9 Seguridad y manejo de residuos B.7.4.9.1 Seguridad Cada reactivo utilizado en estos procedimientos debe ser tratado como un riesgo potencial para la salud y la exposición a estos materiales debe ser minimizada. Cada laboratorio es responsable de mantener un conocimiento de las regulaciones respecto a la manipulación segura de cualquier producto químico usado en este método. Debe ponerse a disposición de todo el personal involucrado en el análisis las hojas de datos de seguridad de los productos químicos. Debe de utilizarse equipo de protección personal apropiado para el manejo de muestras y estándares. Los estándares primarios deberán prepararse en una campana. Cuando el analista maneje altas concentraciones de compuestos tóxicos deberá utilizarse un respirador de gases tóxicos. Las muestras desconocidas pueden ser peligrosas. Manéjelas siempre con precaución extrema. Cuando trabaje con muestras desconocidas o con los reactivos empleados en este método use siempre el equipo de seguridad adecuado como bata de trabajo, lentes de seguridad y guantes apropiados. B.7.4.9.2 Manejo de residuos Es la responsabilidad del laboratorio cumplir con todos los reglamentos federales, estatales, locales y demás disposiciones aplicables referentes al manejo de residuos, particularmente las reglas de identificación, almacenamiento y disposición de residuos peligrosos. B.7.4.10 Referencias EPA Method 350.1 Determination of ammonia nitrogen by semi-automated colorimetry. U.S. Environmental Protection Agency, Environmental Monitoring Systems Laboratory, 1993. B.7.5 MÉTODO PARA LA DESTILACIÓN PRELIMINAR DE MUESTRAS DE AGUA PARA USO Y CONSUMO HUMANO EN LAS QUE SE DETERMINARÁ NITRÓGENO AMONIACAL POR EL MÉTODO DE PRUEBA POR TITULACIÓN ( PUNTO B.7.1, DE ESTE APÉNDICE), POR EL MÉTODO POR ELECTRODO SELECTIVO DE AMONÍACO (PUNTO B.7.2, DE ESTE APÉNDICE) O POR EL MÉTODO CON FENATO (PUNTO B.7.3, DE ESTE APÉNDICE) B.7.5.1 Principio La muestra es amortiguada en un pH de 9.5 unidades de pH con una disolución amortiguadora de borato para disminuir la hidrolisis de cianatos y compuestos orgánicos nitrogenados. Esta es destilada dentro de una disolución de ácido bórico cuando se usa el método por titulación es utilizado o en H2SO4 cuando se usa el método de fenato. El amoníaco destilado puede ser determinado tanto colorimétricamente por el método de fenato o por titulación con H2SO4 estandarizado y una mezcla de indicadores o un medidor de pH. La selección entre el método colorimétrico y los métodos de acidimetría depende de la concentración de amoníaco. El amoníaco en el destilado también puede ser determinado por el método de electrodo selectivo de amoníaco usando H2SO4 0.04 N para atrapar el amoníaco. B.7.5.2 Equipos y materiales Aparato de destilación. Constituido por un matraz de vidrio de borosilicato de 800 a 2 000 mL de capacidad unido a un condensador vertical de tal manera que la punta de salida esté sumergida debajo de la superficie de la disolución ácida que recibe el destilado. Utilizar un dispositivo completamente de vidrio de borosilicato o uno con unidades de condensación construidas de estaño o tubos de aluminio. B.7.5.3 Reactivos y soluciones Ácido sulfúrico 0.04N. Diluir 1 mL de H2SO4 en 1L. Agua libre de amoníaco. Puede ser preparada por intercambio iónico o por métodos de destilación. Para prepararla por intercambio iónico, se debe de pasar agua destilada a través de una columna de intercambio iónico que contiene una resina de intercambio catiónico acídico fuerte mezclada con una resina de intercambio aniónico básica fuerte. Seleccionar las resinar que removerán los compuestos orgánicos que interfieren con la determinación de amoniaco. Algunas resinas de intercambio aniónico tienden a liberar amoniaco. Si esto ocurre, preparar agua libre de amoniaco con una resina de intercambio catiónico acídico fuerte. Regenerar la columna de acuerdo con las instrucciones del fabricante. Verificar el agua esté libre de amoniaco por la posibilidad de obtener un valor alto en el blanco. Para prepararla por destilación, eliminar las trazas de amoniaco en el agua destilada añadiendo 0.1 mL de H2SO4 a 1 L de agua destilada y redestilar. Alternativamente tratar el agua destilada con suficiente agua de bromo o cloro para producir un halógeno residual libre de 2 a 5 mg/L y redestilar después de dejar reposar al menos 1 h. Descartar los primeros 100 mL del destilado. Verificar el agua esté libre de amoniaco por la posibilidad de obtener un valor alto en el blanco. Es muy difícil almacenar agua libre de amoniaco en el laboratorio sin contaminación por amoniaco gaseoso. Sin embargo, si el almacenaje es necesario, guardar en un contenedor de vidrio fuertemente tapado al cual se ha añadido cerca de 10 g de resina de intercambio iónico (preferiblemente una resina de intercambio catiónico acídico fuerte) por litro de agua libre de amoniaco. Para su uso, deje que la resina se asiente y decante el agua libre de amoniaco. Si se produce un blanco alto, reemplace la resina o prepare nueva agua libre de amoniaco. Utilizar agua destilada libre de amoniaco para preparar todos los reactivos y diluciones de las muestras. Agente para neutralización. Hidróxido de sodio NaOH 1N ó ácido sulfúrico H2SO4 1N. Reactivo de descloración. Disolver 3.5 g de tiosulfato de sodio (Na2S2O3 5 H2O) en agua y diluir a 1 L. Preparar una disolución fresca cada semana. Utilizar 1 mL de este reactivo para remover 1mg/L de cloro residual en 500 mL de muestra. Disolución amortiguadora de borato. Añadir 88 mL de una disolución de NaOH 0.1 N a 500 mL aproximadamente de una disolución 0.025 M de tetraborato de sodio (Na2B4O7) (9.5 g Na2B4O7·10 H2O/L) y diluir a 1 L. Disolución indicadora de ácido bórico. Disolver 20 g de H3BO3 en agua y diluir a 1 L B.7.5.4 Procedimiento Preparar el equipo añadiendo 500mL de agua a 20 mL del amortiguador de borato, ajustar el pH a 9.5 unidades de pH con una disolución de NaOH 6N y añadir a un matraz para destilación. Añadir unas cuantas perlas de vidrio y utilizar esta mezcla para purgar con vapor el aparato de destilación hasta que el destilado no muestre trazas de amoniaco. Preparar la muestra usando 500 mL de muestra desclorinada o una porción conocida diluida a 500 mL con agua reactivo. Cuando la concentración de NH3-N es menor a 100 µg/L utilizar un volumen de muestra de 1000 mL. Remover el cloro residual añadiendo al momento del muestreo, reactivo de descloración equivalente al cloro residual presente en la muestra. Si es necesario neutralizar a un pH aproximado de 7 con ácido o base diluido utilizando un medidor de pH. Añadir 25 mL de disolución amortiguadora de borato y ajustar a un pH de 9.5 con NaOH 6 N utilizando un medidor de pH. Para minimizar la contaminación dejar el aparato de destilación ensamblado después de la purga con vapor y justa hasta antes de comenzar la destilación de la muestra. Desconectar el matraz de destilación e inmediatamente transferir la muestra del matraz al aparato de destilación. Destilar a una velocidad de 6 a 10 mL/min con la punta del tubo de salida debajo de la superficie de la disolución acida que recibe el destilado. Recibir el destilado en un matraz Erlenmeyer de 500 mL de capacidad que contiene 50 mL de disolución indicadora de ácido bórico para el método por titulación (punto B.7.1, de este Apéndice). Destilar el amoniaco en 50 mL de H2SO4 0.04 N para el método por electrodo selectivo de amoníaco (punto B.7.2, de este Apéndice) y para el método con fenato (punto B.7.3, de este Apéndice). Recibir al menos 200 mL del destilado. Bajar el receptor de destilación de tal manera que el tubo de salida esté fuera del líquido y continuar la destilación durante dos minutos para limpiar el condensador y el tubo de salida. Diluir a 500 mL con agua reactivo. Cuando el método con fenato es utilizado para determinar el NH3-N de debe neutralizar el destilado con una disolución de NaOH 1N. Posterior a la destilación, determinar el contenido de amoniaco por el método por titulación (punto B.7.1, de este Apéndice), el método por electrodo selectivo de amoníaco (punto B.7.2, de este Apéndice) o por el método con fenato (punto B.7.3, de este Apéndice). B.7.5.5 Referencias Standard methods for examination of water and wastewater. 4500-NH3 B. Preliminary distillation step. B.8 MÉTODO DE PRUEBA PARA LA DETERMINACIÓN DE YODO RESIDUAL LIBRE EN AGUA PARA USO Y CONSUMO HUMANO El yodo elemental no es un componente natural de las aguas naturales. El yodo se puede agregar al agua para uso y consumo humano para su desinfección. El uso de yodo generalmente está restringido a pequeños o remotos sistemas de abastecimiento de agua para uso y consumo donde la facilidad de aplicación, la estabilidad de almacenamiento y la inactividad hacia la materia orgánica son consideraciones importantes. Sin embargo, algunos de los metabolitos derivados de la hidrolisis del yodo elemental, como el ácido hipoyodoso (HOI) o el hipoyodito (OI-), pueden actuar como agente yodinantes, reaccionando con compuestos orgánicos para formar compuestos orgánicos yodados. El yodo se aplica en forma elemental o se produce in situ mediante la adición simultánea de una sal de yoduro y un oxidante adecuado. En este último caso, un exceso de yoduro puede ser mantenido para servir como un reservorio para la producción de yodo, por lo que la determinación del yoduro es necesaria para el control de su uso como desinfectante de agua para uso y consumo humano y con el fin de que la población no este expuesta a concentración innecesariamente altas de yodo. Sin embargo, no existe un método generalmente aceptado para la determinación de cada una de las especies individuales o metabolitos derivados de la hidrolisis de yodo y la mayoría de los métodos analíticos y de campo utilizan el poder oxidante de todas las formas de yodo activo para su determinación y los resultados suelen expresarse como una concentración equivalente de yodo elemental o yodo residual libre. Con la finalidad de proteger la salud de la población es importante que la determinación de los residuales de la desinfección como el yodo se realice en campo directamente in situ en la toma domiciliaria o en la red de distribución, por lo que el uso de métodos de campo (punto B.8.1, de este Apéndice) es necesario. Sin embargo, de ser necesaria la determinación en laboratorio de yodo residual libre en agua para uso y consumo humano tratada con yodo elemental puede realizarse a través de los métodos de titulación amperométrica o colorimétrico de cristal violeta-leuco referidos en el punto B.8.2, de este Apéndice. B.8.1 MÉTODO DE PRUEBA PARA LA DETERMINACIÓN DE YODO RESIDUAL LIBRE EN AGUA PARA USO Y CONSUMO HUMANO EN TOMA DOMICILIARIA O EN RED DE DISTRIBUCIÓN IN SITU POR MEDIO DE KITS COLORIMETRICOS O FOTOMETRICOS B.8.1.1 Principio Existen diversos kits comercialmente disponibles para la determinación de la concentración de yodo residual libre a través de la cuantificación del poder oxidante de todas las formas de yodo activo por medio de métodos colorimétricos o fotométricos. B.8.1.2 Alcance y aplicación Este método es utilizado para la determinación de yodo residual libre a través del uso de kits colorimétricos o fotométricos en muestras de agua para uso y consumo humano colectadas en la toma domiciliaria o en la red de distribución. B.8.1.3 Equipos y materiales Kits colorimétricos o fotométricos para la determinación de yodo residual libre con un intervalo de lectura de incluya 0.2 a 1.5 mg/L. B.8.1.4 Procedimiento B.8.1.4.1 Selección de sitios de muestreo. Para cada sistema formal de abastecimiento, identificar las localidades donde se utilice yodo como desinfectante del agua para uso y consumo humano. B.8.1.4.2 Revisión del equipo de muestreo. Previo al muestreo revisar que el equipo para la determinación de yodo residual libre se encuentre en buen estado y calibrarse de ser necesario. B.8.1.4.3 Monitoreo de Yodo residual libre. Realizar un recorrido en las localidades donde existe yodador para la administración de yodo al sistema de abastecimiento y llevar a cabo la determinación de yodo residual libre directamente en la toma domiciliaria o en una toma de la red de distribución y en el punto de la localidad más alejado del yodador. B.8.1.4.4 Eliminación de interferencias. Para evitar la presencia de interferencia debe retirar cualquier aditamento conectado al grifo (mangueras, etc.). B.8.1.4.5 Eliminación de agua estancada en toma. El agua que se encuentra en la tubería normalmente está estática por lo cual no es representativa de la calidad del agua de la red, por lo que es necesario dejar correr el agua a flujo máximo en el grifo con el fin de purgar la toma y asegurarse que el líquido contenido en la tubería se haya descargado. B.8.1.4.6 Análisis. Determinar el yodo residual libre empleando el kit colorimétrico o fotométrico siguiendo las instrucciones del fabricante. En el formato correspondiente anotar la identificación del punto de muestreo, fecha y hora de muestreo y el resultado de la determinación. B.8.1.4.7 Informe de prueba. La expresión de resultados debe ser mg de yodo residual libre por L con una cifra decimal. B.8.2 MÉTODO DE PRUEBA PARA LA DETERMINACIÓN DE YODO RESIDUAL LIBRE EN AGUA PARA USO Y CONSUMO HUMANO EN LABORATORIO De ser necesaria la determinación en laboratorio de yodo residual libre en agua para uso y consumo humano tratada con yodo elemental puede realizarse utilizando los métodos por titulación amperométrica o colorimétrico de leuco cristal violeta, como los desarrollados en el Standard methods for examination of water and wastewater. 4500-I IODINE, u otras referencias equivalentes. B.9 MÉTODO DE PRUEBA PARA LA DETERMINACIÓN DE pH EN AGUA PARA USO Y CONSUMO HUMANO B.9.1 Definiciones y términos Potencial de hidrógeno (pH), al logaritmo negativo de la concentración del ion hidrógeno en una disolución acuosa o el logaritmo del recíproco de la concentración de iones hidrógeno. B.9.2 Símbolos y términos abreviados pH potencial hidrógeno. B.9.3 Principio Se basa en la determinación de la actividad de iones hidrógeno (H+) medidos en un potenciómetro usando un electrodo de vidrio y otro de referencia o un electrodo combinado. La fuerza electromotriz producida por el sistema de electrodo(s) es proporcional al pH de la disolución problema (se usa el término de iones H+ para mayor claridad reconociendo que en realidad existe en su forma hidratada, el ion hidronio H3O+). B.9.4 Alcance y aplicación Este método es utilizado para la determinación de pH en muestras de agua para uso y consumo humano a través de un potenciómetro en laboratorio. B.9.5 Equipos y materiales Sólo se mencionan los equipos y materiales que no son de uso común en el laboratorio analítico. Todo el material volumétrico utilizado debe ser clase A o estar verificada su calibración. Agitador de vidrio Agitador magnético y barra magnética recubierta con plástico inerte o sonda de agitación o agitador de vidrio. Baño María de temperatura constante o celda para ajustar temperatura. Electrodo combinado para pH. Frascos con tapón para contener los patrones de pH y las muestras. Matraces Erlenmeyer de 125 mL. Papel para secar el electrodo. Piceta Potenciómetro para medir pH. Termómetro de líquido en vidrio. Tubos de ensayo con rosca para tapones, para contener las muestras. B.9.6 Reactivos y soluciones Los reactivos que requiere el método deben ser tipo ACS grado reactivo a menos que otra cosa se indique. Agua grado reactivo Soluciones amortiguadoras de pH 4 ,7 y 10, con certificado de análisis trazable a patrones nacionales o internacionales. B.9.7 Procedimiento B.9.7.1 Procedimiento analítico Colocar los patrones y las muestras en recipientes tapados y sumergirlos, en baño María a temperatura constante, para alcanzar la temperatura de 25 °C, puede utilizarse un compensador automático de temperatura. Calibrar el potenciómetro siguiendo las instrucciones del fabricante y después de cada medición, lavar el electrodo con agua reactivo tipo I, quitar el exceso con papel secante que no deje residuos, evitar colocar el bulbo sensor y/o friccionar la superficie de electrodo. Verificar que las lecturas de los patrones den valores dentro del intervalo de ± 0.1 unidades de pH a 25 °C de acuerdo a las especificaciones de su certificado. Verificar que la eficiencia electromotriz sea a 95%. Previamente a las mediciones homogenizar y expulsar lo más posible el CO2 de las muestras, agitar cuidadosamente con un agitador de vidrio o con agitación automática durante 10 segundos. Para la medición en muestras amortiguadas de potencia iónica fuerte, para acondicionar electrodos, después de la limpieza, sumergir en la muestra por un minuto, secar y medir pH en otras dos porciones de muestras. Para muestras diluidas, soluciones pobremente amortiguadas, se recomienda remojar y enjuagar el electrodo previamente en tres o cuatro porciones de muestra y tomar lecturas de pH en otras dos o tres porciones de muestra. B.9.7.2 Análisis de datos y cálculos Obtener la lectura directa de las muestras y reportar el promedio a 25 °C. B.9.7.3 Informe de prueba. La expresión de resultados debe ser en unidades de pH y con una cifra decimal. B.9.8 Referencias Standard methods for examination of water and wastewater. 4500-H+ B. Electrometric Method. B.10 MÉTODO DE PRUEBA PARA LA DETERMINACIÓN DE DUREZA TOTAL EN AGUA PARA USO Y CONSUMO HUMANO B.10.1 Símbolos y términos abreviados CaCO3 carbonato de Calcio EDTA Ácido etilendiaminotetraacético NaCl cloruro de sodio B.10.2 Principio Este método emplea como indicador el compuesto químico denominado negro de eriocromo T, el cual al ser agregado a una disolución que contenga iones calcio y magnesio, reacciona formando complejos de un color vino. Después se adiciona la disolución de EDTA que remueve los iones calcio y magnesio de los complejos coloridos formando complejos solubles. Cuando ha sido agregada suficiente disolución EDTA, para liberar todos los iones calcio y magnesio, el indicador regresa a su color azul original. El término original para la dureza en agua, se refiere a la medida de la capacidad del agua para precipitar jabón. Este fenómeno se produce principalmente por los iones de calcio y magnesio, aunque pueden intervenir otros metales polivalentes, tales como fierro, zinc, aluminio, manganeso y estroncio, además de los iones hidrógeno. La presencia misma de estos iones en aguas naturales origina que la definición de dureza represente la concentración total de magnesio y calcio únicamente, indicando su cálculo final en concentración de carbonato de calcio. B.10.3 Alcance y aplicación Este método es utilizado para la determinación de la dureza total en muestras de agua para uso y consumo humano a través del método por titulación con EDTA. B.10.4 Equipos y materiales Sólo se mencionan los equipos y materiales que no son de uso común en el laboratorio analítico. Todo el material volumétrico utilizado debe ser clase A o estar verificada su calibración. Balanza analítica con sensibilidad de ± 0.1 mg Buretas graduadas calibradas de 25 y 50 mL Matraz Erlenmeyer de 500 mL, 250 mL y 125 mL Pinza para bureta Pipetas graduadas de 10 mL o dosificador Pipetas volumétricas de 50 mL o dosificador Potenciómetro para medir pH Soporte universal Vaso de precipitados de 100 mL B.10.5 Reactivos y soluciones Los reactivos que requiere el método deben ser tipo ACS grado reactivo a menos que otra cosa se indique. Agua grado reactivo Disolución amortiguadora, disolver 1.179 g de la sal disódica dihidratada de EDTA y 780 mg de sulfato de magnesio (MgSO4 7H2O) o 644 mg de cloruro de magnesio (MgCl2 6H2O) en 50 mL de agua. También puede prepararse disolviendo 16.9 g de Cloruro de Amonio (NH4Cl) en 143 mL de hidróxido de amonio (NH4OH), adicionar 1.25 g de EDTA y disolver en 250 mL de agua. Guardar la disolución en un recipiente de vidrio resistente, o de plástico perfectamente tapado. Desechar en cuanto en la porción de muestra utilizada no produzca un pH de 10±0.1. Indicador, mezclar 0.5 g de eriocromo negro T, sal sódica del ácido 1-hidroxi-2-naftilazo-5-nitro-2-naftol-4- sulfónico y 100g de NaCl, evitar contacto con el aire. En caso de que el punto final de la titulación no se distinga, es necesario obtener nueva mezcla indicadora. Disolución patrón titulante de EDTA 0,01 M, pesar 3.723 g de la sal disódica del ácido etilen diamino tetraacético, Na2H2C10H12O8N2·2H2O, disolver en agua reactivo y diluir a 1 litro. Verificar su concentración titulando contra disolución de carbonato de calcio patrón. Almacenar en recipiente de polietileno o vidrio pyrex. Disolución patrón de CaCO3, Pesar 1.0 g de carbonato de calcio anhidro, colocarlo en un matraz de 500mL, agregar HCl 1+1, hasta que todo el carbonato sea disuelto, agregar 200mL de agua destilada y hervir durante algunos minutos para desalojar el CO2, enfriar y agregar unas gotas de disolución indicadora de rojo de metilo, ajustar al color anaranjado, agregando unas gotas de disolución de NH4OH 3N. La disolución patrón es equivalente a 1.000 mg de CaCO3 por cada mL. B.10.6 Procedimiento B.10.6.1 Valoración de la disolución de EDTA Medir 10 mL de la disolución estándar de carbonato de calcio, por triplicado en un matraz Erlenmeyer de 250 mL. Agregar de 1 a 2mL de la disolución amortiguadora (se deberá tener pH de 10.0±0.1) Agregar la cantidad adecuada del indicador en polvo de negro de eriocromo T. Titular con la disolución de EDTA, hasta el vire de violeta a azul. Calcular el factor F, de titulación, con la fórmula siguiente:  B.10.6.2 Procedimiento analítico Medir una alícuota de 50 mL de muestra, transferir a un matraz de 200 mL. Adicionar 1 a 2 mL de disolución amortiguadora y agitar. La titulación debe efectuarse a un pH específico de 10.0±0.1. Verificar y registrar la lectura de pH. Adicionar una adecuada cantidad de indicador, de tal manera que se perciba el vire característico de color violeta a color azul. Si la muestra da inmediatamente un color azul, significa que no tiene dureza. Titular la muestra agregando gota a gota la disolución titulante de EDTA, agitando continuamente hasta el punto final en que se produce el cambio de color rojo vino a azul. Los mL gastados de disolución de EDTA deben ser menores a 15 mL en caso contrario diluir la muestra. En caso de que no se aprecie el vire, será necesario usar indicador recién hecho y/o agregar inhibidor y/o aplicar digestión, de acuerdo a la supuesta interferencia presente. Puede ser necesario seguir procesos de eliminación de interferencias de acuerdo a lo indicado anteriormente. B.10.6.3 Análisis de datos y cálculos Calcular la dureza total utilizando la siguiente fórmula: 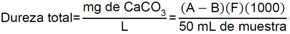 En donde: A mL de disolución de EDTA empleados en la titulación de la muestra B mL de disolución de EDTA empleados en la titulación del blanco, cuando se utiliza alícuota de muestra llevada a 50 mL con agua reactivo F equivalente a mg de CaCO3 por mL de disolución de EDTA, obtenido en la valoración B.10.6.4 Informe de prueba La expresión de resultados debe ser en 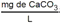 y con dos cifras decimales. y con dos cifras decimales. B.10.7 Interferencias Algunos iones metálicos interfieren causando un vire difícil de percibir o por consumo estequiométrico de EDTA. La sal de magnesio del ácido 1, 2-ciclohexanodiamina tetraacético (MgCDTA), es selectivamente compleja a los metales pesados, liberando Mg en la muestra, y puede ser usado en lugar de los inhibidores. Usar cuando el Mg sustituido por los metales no contribuya significativamente a la dureza total. Agregar 250 mg del MgCDTA por cada 100 mL de muestra y disolver completamente antes de adicionar la disolución amortiguadora. Las interferencias también se reducen con el uso de inhibidores, que funcionan como agentes complejantes (Tabla B.10-1, de este Apéndice).
Para la mayoría de las aguas no son necesarios estos agentes complejantes. Si es necesario usarlos de acuerdo a tabla anterior. 1) Inhibidor I: Ajustar las muestras acidas a pH 6 o más alto con disolución amortiguadora o con NaOH 0.01 N. Agregar 250 mg de cianuro de sodio en polvo. Adicionar suficiente disolución amortiguadora para alcanzar pH de 10±0.1 (tomar precaución en su uso, puesto que el cianuro es extremadamente venenoso). Tomar especial precaución para prevenir su contacto con ácidos, lo cual puede liberar al volátil y venenoso cianuro de hidrógeno. 2) Inhibidor II: Disolver 5.0 g de sulfuro de sodio nonahidratado, NaS·9H2O, o 3.7 g de NaS·5H2O en 100 mL de agua reactivo. Excluir el aire con un tapón de hule. Este inhibidor se deteriora por oxidación con aire. Se produce una precipitación de sulfuro que oscurece el punto final cuando una apreciable cantidad de metales está presente. La materia orgánica o suspendida puede interferir con el punto final. Eliminar esta interferencia evaporando la muestra a sequedad sobre baño de vapor y calentar en una mufla a 500°C hasta que la materia orgánica este oxidada. Disolver el residuo en 20 mL de HCl 1 N, neutralizar a pH 7 con NaOH 1N y llevar a 50 mL con agua reactivo, enfriar a temperatura ambiente y continuar con el procedimiento general. Llevar a cabo las titulaciones a temperatura ambiente. Cuándo la muestra está cerca de la congelación el vire es muy lento. En agua caliente el indicador se descompone. Para evitar la precipitación de CaCO3, la cual produce bajos resultados se recomienda: 1) Titular dentro de un tiempo de 5 minutos. 2) Usar la dilución mencionada en tabla anterior. Si la precipitación es apreciable, todavía, usar las recomendaciones 3 y 4, citadas en este punto. Usar pequeñas muestras contribuye a un error sistemático debido al error de lectura de la bureta. 3) Si la dureza aproximada es conocida o se determinó por una titulación previa, agregar el 90% o más del titulante antes de ajustar el pH con la disolución amortiguadora. 4) Acidificar la muestra con H2SO4 0.02 N, y agitar por 2 minutos para expulsar el CO2 antes del ajuste de pH. Determine alcalinidad para que indique la cantidad de ácido agregado. B.10.8 Referencias Standard methods for examination of water and wastewater. 2340 C EDTA Titrimetric Method. B.11 MÉTODO DE PRUEBA PARA LA DETERMINACIÓN DE SULFATO EN AGUA PARA USO Y CONSUMO HUMANO Para la determinación de sulfato en agua para uso y consumo humano para el cumplimiento de esta Norma, se podrá utilizar indistintamente el: · método de prueba turbidimétrico para la determinación de sulfato en agua para uso y consumo humano (punto B.11.1, de este Apéndice). · método de prueba por colorimetría automatizada para la determinación de sulfato en agua para uso y consumo humano (punto B.11.2, de este Apéndice). B.11.1 MÉTODO DE PRUEBA TURBIDIMÉTRICO PARA LA DETERMINACIÓN DE SULFATO EN AGUA PARA USO Y CONSUMO HUMANO B.11.1.1 Principio El ión sulfato es precipitado en medio ácido con cloruro de bario, BaCl2, de tal manera que se forman cristales de sulfato de bario de tamaño uniforme, produciéndose una turbidez medible y proporcional a la concentración de sulfato. La absorbancia producida por la suspensión de sulfato de bario se mide en el espectrofotómetro a 420 nm. Las aguas naturales normalmente contienen concentraciones de sulfato que se pueden presentar en intervalos variables. Pero son posibles las contaminaciones causadas por los drenajes de minas que contienen piritas. Por otro lado se sabe que el sulfato de calcio y magnesio en concentraciones mayores de 250 ppm ejercen una acción catártica, por eso es importante saber el contenido de sulfato. B.11.1.2 Alcance y aplicación Este método es utilizado para la determinación de sulfato en muestras de agua para uso y consumo humano a través del método turbidimétrico, que es aplicable en un intervalo de 1 a 40 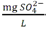 B.11.1.3 Equipos y materiales Agitador y barras magnéticas. Balanza analítica con sensibilidad de ± 0.1 mg. Bureta de 100 mL. Celdas de vidrio o cuarzo de 2.5 a 10 cm de paso de luz. Cuchara con capacidad de 0.2 a 0.3 mL, para medir el cloruro de bario. Cronómetro Espectrofotómetro para leer a 420 nm. Pipetas volumétricas de 5 y 20 mL. Pipeta volumétrica de 100 mL. Vaso de precipitados o matraz Erlenmeyer de 250 mL. B.11.1.4 Reactivos y soluciones Agua reactivo tipo I Cloruro de bario (BaCl2), en cristales. Reactivo acondicionante, obtener una disolución que contenga 30 mL de HCl concentrado, 30 0mL de agua reactivo tipo I, 100 mL de alcohol etílico o isopropílico al 95% y 75g de NaCl, disolver y agregar 50mL de glicerina. Disolución patrón de sulfato, que contenga 0.10 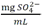 , disolver 0.1479 g de Na2SO4 anhidro, en agua tipo I y aforar a un 1L. Se puede utilizar una disolución comercial de sulfato con certificado de análisis trazable a patrones nacionales o internacionales. , disolver 0.1479 g de Na2SO4 anhidro, en agua tipo I y aforar a un 1L. Se puede utilizar una disolución comercial de sulfato con certificado de análisis trazable a patrones nacionales o internacionales. B.11.1.5 Procedimiento B.11.1.5.1 Corrección por turbidez o color de un blanco de reactivos para patrones y muestras Realizar una curva de comparación. Medir 100 mL de muestra, o una alícuota adecuada diluida a 100mL en un recipiente de 250mL y colocar la barra de agitación. Agregar 5 mL de reactivo acondicionante, mezclar con agitación durante 1 minuto a velocidad constante. Tomar la lectura de turbidez a 420nm. B.11.1.5.2 Medición de la turbiedad del sulfato de bario Agregar a patrones y muestras una cucharilla llena de cristales de cloruro de bario y agitar durante 60 segundos. Tomar la lectura de sulfato a la absorbancia de 420 nm. B.11.1.5.3 Análisis de datos y cálculos Obtener la diferencia de las absorbancias obtenidas anteriormente, y sustituirla en la ecuación de la curva de calibración ajustada por mínimos cuadrados considerando lo siguiente: 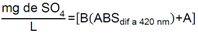 m y b son las constantes obtenidas con el ajuste de mínimos cuadrados para la curva de calibración. 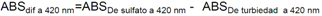 y es la diferencia de absorbancias por sulfato y por turbiedad y es la diferencia de absorbancias por sulfato y por turbiedad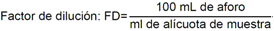 B.11.1.5.4 Informe de prueba La expresión de resultados debe ser en  y con dos cifras decimales. y con dos cifras decimales. B.11.1.6 Referencias Standard methods for examination of water and wastewater. 427 C. Turbidimetric Method. B.11.2 MÉTODO DE PRUEBA POR COLORIMETRÍA AUTOMATIZADA PARA LA DETERMINACIÓN DE SULFATO EN AGUA PARA USO Y CONSUMO HUMANO B.11.2.1 Definiciones y términos Las definiciones presentadas en esta sección son específicas para este método, pero han sido conformadas para que sean lo más posible de uso común. Bitácora, al cuaderno de laboratorio debidamente foliado e identificado, en el cual los analistas anotan todos los datos de los procedimientos que siguen durante el análisis de una o varias muestras, así como toda la información pertinente y relevante a su trabajo en el laboratorio. Blanco, al agua reactivo o matriz equivalente a la que no se le aplica ninguna parte del procedimiento analítico y sirve para evaluar la señal de fondo. Blanco analítico o de reactivos, al agua reactivo o matriz equivalente que no contiene, por adición deliberada, la presencia de ningún analito o sustancia por determinar, pero que contiene los mismos disolventes, reactivos y se somete al mismo procedimiento analítico que la muestra problema. Calibración, al conjunto de operaciones que establecen, bajo condiciones específicas, la relación entre los valores de una magnitud indicados por un instrumento o sistema de medición, o los valores representados por una medida materializada y los valores correspondientes de la magnitud, realizados por los patrones, efectuando una corrección del instrumento de medición para llevarlo a las condiciones iniciales de funcionamiento. Desviación estándar experimental, para una serie de mediciones del mismo mensurando, es la magnitud que caracteriza la dispersión de los resultados. Disolución patrón, a la disolución de concentración conocida preparada a partir de un patrón primario. Disolución estándar, a la disolución de concentración conocida preparada a partir de un patrón primario. Disolución madre, corresponde a la disolución de máxima concentración en un análisis. Es a partir de esta disolución que se preparan las disoluciones de trabajo. Exactitud, a la proximidad de concordancia entre el resultado de una medición y un valor verdadero del mesurado. Límite de detección del método (LDM), a la concentración mínima del analito que puede detectarse con un nivel de confianza predeterminado. Para efectos de esta Norma, el nivel de confianza es del 99%. Este límite de detección generalmente se logra por analistas experimentados con equipo bien calibrado y bajo condiciones no rutinarias. Límite práctico de cuantificación (LPC), a la concentración mínima del analito que puede determinarse con un nivel de confianza predeterminado en condiciones rutinarias de operación. Este límite puede establecerse entre 5 a 10 veces el LDM. Medición, al conjunto de operaciones que tiene por objeto determinar el valor de la magnitud. Mensurando, a la magnitud particular sujeta a medición. Material de referencia, al material o sustancia en el cual uno o más valores de sus propiedades son suficientemente homogéneos y bien definidos, para ser utilizadas para la calibración de aparatos, la evaluación de un método de medición o para asignar valores a los materiales. Material de referencia certificado, material de referencia, acompañado de un certificado, en el cual uno o más valores de las propiedades están certificados por un procedimiento que establece la trazabilidad a una realización exacta de la unidad en la cual se expresa los valores de la propiedad, y en el que cada valor certificado se acompaña de una incertidumbre con un nivel declarado de confianza. Muestra de control de calidad (MCC), a la muestra que contiene todos o un subgrupo de los analitos del método a concentraciones conocidas. Las MCC se obtienen de una fuente externa al laboratorio o se preparan de una fuente de estándares diferentes de la fuente de los estándares de calibración. Parámetro, a la variable que se utiliza como referencia para determinar la calidad del agua. Patrón de medición, al material de referencia, instrumento de medición, medida materializada o sistema de medición destinado a definir, realizar, conservar o reproducir una unidad o uno o más valores de una magnitud para utilizarse como referencia. Patrón nacional de medición, al patrón reconocido por una decisión nacional en un país, que sirve de base para asignar valores a otros patrones de la magnitud concerniente. Patrón primario, al patrón que es designado o reconocido ampliamente como un patrón que tiene las más altas cualidades metrológicas y cuyo valor es aceptado sin referencia a otros patrones de la misma magnitud. Patrón secundario, al patrón cuyo valor es establecido por comparación con un patrón primario de la misma magnitud. Patrón de referencia, al patrón, en general de la más alta calidad metrológica disponible en un lugar dado, o en una organización determinada de la cual se derivan las mediciones realizadas en dicho lugar. Patrón de trabajo, al patrón que es usado rutinariamente para calibrar o controlar las medidas materializadas, instrumentos de medición o los materiales de referencia. Precisión, al grado de concordancia entre resultados analíticos individuales cuando el procedimiento analítico se aplica repetidamente a diferentes alícuotas o porciones de una muestra homogénea. Usualmente se expresa en términos del intervalo de confianza o incertidumbre. B.11.2.2 Símbolos y términos abreviados AMT azul de metiltimol LDM límite de detección del método LPC límite práctico de cuantificación 8 botón de inicio de análisis B.11.2.3 Principio La muestra se hace pasar por una columna de intercambio catiónico para eliminar iones multivalentes. Después, el sulfato en la muestra reacciona con un complejo de bario- azul de metiltimol (Ba-AMT) a un pH entre 2.5 y 3.0 para obtener sulfato de bario (BaSO4) y azul de metiltimol libre (AMT) (Fig. B.11.2-1, de este Apéndice). Posteriormente se eleva el pH de la disolución a un valor entre 12.5 y 13.0. A este pH, el complejo [Bario (AMT)] es azul (l máx=610 nm), mientras que el AMT libre es gris (lmáx =460 nm). Dado que las concentraciones molares de bario y de AMT son aproximadamente iguales, la concentración de sulfato en la muestra es directamente proporcional a la concentración de AMT libre (siempre y cuando la concentración de sulfato no exceda la concentración del complejo Bario-AMT) el cual se mide a 460 nm. La curva de calibración de sulfato no se ajusta a un modelo de primer orden, sino a uno de tercero. Se ha sugerido que esto se debe a la formación de complejos binucleares de Bario-AMT y a las impurezas que contiene el AMT comercial. 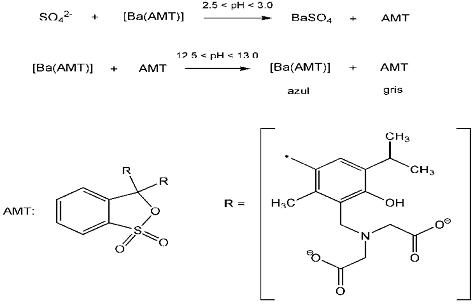 Fig. B.11.2-1. Reacción entre el Ion sulfato y el complejo bario azul de metiltimol (Ba-AMT). B.11.2.4 Alcance y aplicación Este método es utilizado para la determinación de sulfato en un intervalo de 5 a 200 mg/L en agua para uso y consumo humano. El intervalo de trabajo se puede extender hacia concentraciones más altas mediante la dilución de las muestras. B.11.2.5 Equipos y materiales Las referencias a marcas específicas y números de catálogo se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden representar especificaciones adecuadas para los artículos. Sólo se mencionan los equipos y materiales que no son de uso común en el laboratorio analítico. Todo el material volumétrico utilizado debe ser clase A o estar verificada su calibración. Analizador automático, que incluye: bomba peristáltica de canales múltiples; automuestreador; detector fotométrico con celda de flujo; sistema de recopilación de datos: Computadora con el programa requerido instalado; y cartucho configurado para el análisis de sulfato (Fig. B.11.2-2, de este Apéndice). Si se usa el equipo en la modalidad de FIA, el equipo debe contar con la válvula apropiada. 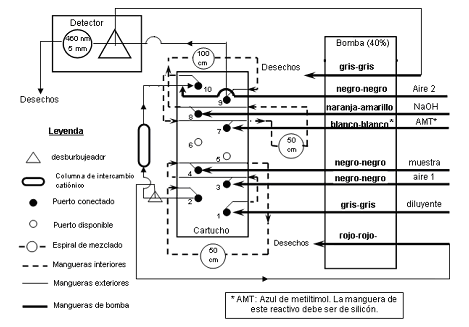 Fig. B.11.2-2. Configuración del cartucho para el análisis de sulfato en el intervalo de 5.0-200 mg/L Balanza Columna de intercambio catiónico Embudos de filtración Material de vidrio lavado de acuerdo al instructivo de control de calidad. Papel filtro de tamaño de poro de 2 µm o menor (Whatman 2 o equivalente). Potenciómetro o tiras reactivas indicadoras de pH. B.11.2.6 Reactivos y soluciones Las referencias a marcas específicas y números de catálogo se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden representar especificaciones adecuadas para los artículos. Sólo se mencionan los equipos y materiales que no son de uso común en el laboratorio analítico. Todo el material volumétrico utilizado debe ser clase A o estar verificada su calibración. Los reactivos que requiere el método deben ser tipo ACS grado reactivo a menos que otra cosa se indique. Los reactivos empleados en este método se indican en la Tabla B.11.2-1, de este Apéndice.
B.11.2.7 Procedimiento B.11.2.7.1 Preparación de soluciones Para mejores resultados, filtre con membrana de 0.45 µm todas las disoluciones de los reactivos preparados antes de usarlos. B.11.2.7.1.1 Ácido clorhídrico 1N (1L). Mientras agita, añadir cuidadosamente 83.3 mL de ácido clorhídrico concentrado a aproximadamente 800 mL de agua desionizada. Al mezclar ácido clorhídrico concentrado con agua se libera una gran cantidad de calor. Tome las precauciones necesarias. Enfriar la disolución a temperatura ambiente, diluir a 1 000 mL con agua desionizada y mezclar bien. B.11.2.7.1.2 Agua Debe cumplir con las especificaciones de agua ASTM tipo II. Es recomendable utilizar agua desionizada que además cumpla las características ASTM tipo II para el uso del equipo FIA. Adicionalmente se puede hacer un tratamiento de desgasificación del agua a utilizar si el analista lo considera prudente con base en el ruido instrumental obtenido en el equipo. (Se sugiere realizar con ruido instrumental > 2000 µuA). Para ello escoger alguna técnica: Someter el agua a un vacío fuerte durante 15 a 20 minutos; agitar magnéticamente o someta a un ultrasonido el agua por 20 minutos; purgar el agua con nitrógeno gaseoso (u otro gas inerte) con ayuda de un material poroso por 5 minutos; o hervir el agua en un matraz Erlenmeyer por 15 minutos y dejar enfriar a temperatura ambiente cubriendo la boca del matraz con un vaso de precipitados invertido. Una vez desgasificada el agua, guárdela en un contenedor cerrado para evitar que absorba gases atmosféricos. B.11.2.7.1.3 Disolución de arranque / disolución de lavado. Esta disolución se usa para ambos propósitos. Añadir 0.5mL de Brij-35 a 500 mL de agua desionizada. Mezclar suavemente. B.11.2.7.1.4 Disolución de azul de metiltimol de trabajo (500mL). 7.2.8.1. Tomar 500 mL de la disolución concentrada de azul de metiltimol y añadir 2.5 mL de Brij-35. Mezclar suavemente. Ajustar el pH de esta disolución a 2.62.7 con NaOH 0.18 N o HCL 1 N. Preparar esta disolución fresca cada día que se analice un lote de muestras. B.11.2.7.1.5 Disolución de trabajo de hidróxido de sodio 0.18 N (500 mL). Añadir 9 mL de la disolución de hidróxido de sodio 10 N a aproximadamente 400 mL de agua desionizada. Llevar a un volumen final de 500mL con agua desionizada y mezclar bien. Guardar esta disolución en frasco de polietileno. B.11.2.7.1.6 Disolución madre de 10 000 mg/L sulfato (SO42-). Materiales de referencia. Pesar con precisión 1.4787g de sulfato de sodio anhidro (Na2SO4) secado previamente a 105 ± 3 °C y que cuente con la pureza necesaria para ser considerado como material de referencia y el certificado que lo demuestre. Disolver en aproximadamente 80mL de agua desionizada dentro de un matraz aforado. Diluir hasta 100 mL con agua desionizada. Guardar en frasco de polietileno y preservar con 3 gotas de cloroformo. Esta disolución es estable por seis meses si se almacena en refrigeración a 4 °C. B.11.2.7.1.7 Disolución concentrada de azul de metiltimol (AMT) (1L). Para obtener mejores resultados este reactivo se prepara al menos 24 horas antes de usarlo. Disolver 0.236 g de sal sódica de azul de metiltimol en 50 mL de la disolución de cloruro de bario dentro de un matraz volumétrico de 1L. Añadir 142 mL de agua desionizada y 8 mL de ácido clorhídrico 1 N. Llevar a un volumen de 1L con etanol. Agitar magnéticamente la disolución por una hora para desgasificarla. Añadir más etanol para llevar nuevamente a un volumen de 1 L. Filtrar con membrana de 0.45 µm. Guardar en frasco de vidrio ámbar. Esta disolución es estable hasta por 2 semanas si se guarda en frasco ámbar y a 4 °C. B.11.2.7.1.8 Disolución concentrada de cloruro de bario (1L). Disolver 1.526 g de cloruro de bario dihidratado en aproximadamente 800mL de agua desionizada. Llevar a un volumen final de 1 000 mL con agua desionizada y mezcle bien. B.11.2.7.1.9 Disolución concentrada de hidróxido de sodio 10N (100mL). Mientras agita, añadir cuidadosamente 40 g de hidróxido de sodio a aproximadamente 70mL de agua desionizada. Mezclar hidróxido de sodio con agua libera una gran cantidad de calor. Tome las precauciones necesarias. Agitar hasta disolver el sólido. Dejar enfriar a temperatura ambiente, diluir hasta 100 mL con agua desionizada y mezclar bien. Guardar esta disolución en frasco de polietileno. Prepare esta disolución mensualmente. B.11.2.7.1.10 Disolución Diluyente (1L). Diluir 0.375 mL de la disolución estándar de 1 000 mg/L de SO42- en aproximadamente 800 mL de agua desionizada. Añadir 4 gotas de Brij-35 y llevar a 1 000 mL con agua desionizada. Mezclar suavemente. Preparar esta disolución fresca cada día que se analice un lote de muestras. B.11.2.7.2 Recolección, preservación y almacenamiento de muestras Las muestras se colectan en envases de vidrio o plástico limpios. La cantidad de muestra debe ser suficiente para garantizar que se tenga una muestra representativa, que permita el análisis de replicados si es necesario y que minimice la cantidad de desechos generados. Se sugiere tomar un volumen de muestra entre 50 y 100 mL. Las muestras deben mantenerse a 4 °C y analizarse lo más pronto posible a partir de su recolección. El tiempo máximo previo al análisis es de 28 días. B.11.2.7.3 Preparación de muestras de agua Tomar un volumen suficiente de muestra para filtrarlo y recuperar un volumen de filtrado de al menos 9 mL. Generalmente un volumen de 20 mL de muestra es suficiente. Para muestras considerablemente turbias este volumen puede ser mayor. Comenzar revisando la lista de analistas y ordene iniciando con las muestras potables y al final las residuales para evitar una posible contaminación de las muestras. En el caso que la lista de analistas presente muestras también para el análisis de Fluoruros ordene primero las potables de fluoruro, seguido por potables de sulfato, residuales de fluoruro y finalmente residuales de sulfato, esto porque es posible analizar en el equipo simultáneamente estas químicas. Filtrar las muestras con papel filtro de poro mediano. Un tamaño de poro de 2.5 µm es suficiente, aunque pude usar un tamaño de poro más pequeño (Equivalente a Alhstom N° 2). Si las muestras filtradas aún presentan coloración, adicionar una pizca de carbón activado, agite y fíltrelas nuevamente. Verificar el pH de las muestras con tiras reactivas indicadoras de pH o con un potenciómetro. Neutralice las muestras que tengan un pH menor a 2 utilizando una disolución concentrada de NaOH. Asegúrese que el pH ácido no se debe a una preservación errónea para este parámetro con ácido sulfúrico. Transferir el líquido resultante a viales de vidrio de 9mL de capacidad para su análisis. B.11.2.7.4 Pruebas semicuantitativas para sulfato Para saber anticipadamente si la concentración de sulfato en las muestras se encuentra por arriba del límite superior del intervalo de trabajo se realiza el siguiente ensayo semicuantitativo. Esta prueba puede hacerse para las muestras que se presuma se cuantifiquen en la zona alta del IT o bien salgan de éste. Esto queda a criterio del analista y del técnico de laboratorio. Tomar aproximadamente 20 mL de cada muestra y adicione a cada una pizca de cloruro de bario sólido y agite suavemente. Haga lo mismo con una alícuota del mismo volumen del estándar de 200 mg/L. Si la muestra contiene sulfato, se genera una turbiedad blanca debido a la formación de sulfato de bario (BaSO4). A mayor concentración del Ion sulfato, la turbiedad es mayor. Comparando las muestras con el estándar de 200 mg/L, se obtiene una buena aproximación para saber si la concentración de sulfato en la muestra es mayor a 200 mg/L y por lo tanto requiere ser diluida. Realice las diluciones necesarias con agua desionizada. B.11.2.7.5 Preparación del autoanalizador Flow Solution IV Configurar el equipo como lo indica el Manual del Fabricante y asegurarse de que todos los módulos estén encendidos. Verificar que las conexiones de las mangueras estén configuradas en el cartucho de análisis como muestra el diagrama de la Fig B.11.2-2, de este Apéndice. No instale la columna de intercambio catiónico en esta parte del procedimiento. Se debe instalar con la bomba en funcionamiento para evitar la entrada de aire en ella. Asegurarse que se tiene instalado el filtro de 460 nm en el detector. Llenar el depósito de enjuague del automuestreador con agua desionizada. B.11.2.7.6 Estabilización del equipo Abrir el programa del equipo, cuyo icono de acceso se encuentra en el escritorio de la computadora. Asegúrese que al hacer esto la cánula del automuestreador se introduzca en la cavidad de enjuague. Conectar las mangueras del NaOH 0.18 N y del diluyente a un recipiente con agua destilada y la manguera del reactivo de azul de metiltimol a un recipiente con disolución de arranque/lavado. Asegure las grapas de todas las mangueras en la bomba presionándolas hacia abajo hasta que escuche un "clic" y levante todas las palancas correspondientes para presionar las mangueras contra los rodillos de la bomba peristáltica. Accione la bomba a una velocidad de 40% permitiendo que el agua y la disolución de arranque fluyan por todo el sistema. Asegurarse de que no existan fugas en las conexiones, que no haya mangueras presionadas y que los flujos en las mangueras sean constantes. B.11.2.7.7 Verificación de la línea base Crear o cargar un método apropiado para el análisis de SO42- en el programa del equipo. Consultar el Manual del Fabricante del programa del equipo para mayor referencia. Crear una secuencia que incluya las muestras que conforman el lote y los controles de calidad. Consultar el Manual del Fabricante del programa del equipo para mayor referencia. Seleccionar la opción Collect Data (o su equivalente) del menú de la ventana principal del programa del equipo. Introduzca el nombre y clave del analista, seleccione el método y la secuencia a usarse en el análisis e indique un nombre y una ruta para guardar los resultados del análisis. Comenzar el monitoreo de la línea base presionando el botón (o su equivalente) ubicado en el margen izquierdo de la pantalla de obtención de datos. Monitorear la línea base por unos minutos. Su magnitud no debe sobrepasar las 2000 µuA y su deriva (desplazamiento hacia arriba o hacia abajo) no debe ser mayor a las 500 µuA en 300 segundos. Si observa fluctuaciones grandes en la línea base o una deriva continua probablemente se deba a la presencia de burbujas en la celda de flujo. Extraer estas burbujas tomando la manguera a la salida del flujo de la celda y forme un bucle con ella. Presionar con los dedos el punto donde se sobreponen las dos secciones de la manguera y tire ligeramente mientras presiona, sosteniendo con la otra mano la porción de la manguera más próxima a la celda de flujo. Observar si se estabiliza la línea base. Repetir esta operación hasta que la línea base sea estable. Conectar todas las mangueras a los recipientes de los reactivos correspondientes y verifique nuevamente las condiciones de la línea base. B.11.2.7.8 Preparación de la columna de intercambio catiónico La columna de intercambio catiónico debe prepararse cada vez que se realice un lote. No instale la columna de intercambio catiónico en esta parte del procedimiento. Se debe instalar con la bomba en funcionamiento para evitar la entrada de aire en ella. Tomar aproximadamente 0.5 g de resina de intercambio catiónico Bio-Rex 70 (forma sódica) y lavarla varias veces en un vaso de precipitados de 100 mL con agua desionizada. Decantar cada vez las partículas finas junto con el sobrenadante. Añadir 100 mL de agua desionizada y someter a un ultrasonido por 20 minutos. Desconectar una de las tuercas en los extremos de la columna de polipropileno de la junta de ¼-28 (Fig. B.11.2-3, de este Apéndice). Con cuidado retire el tapón poroso o frit (No. de parte A000947). El otro extremo de la columna debe estar cerrado con el tapón poroso, la tuerca y la junta correspondientes. 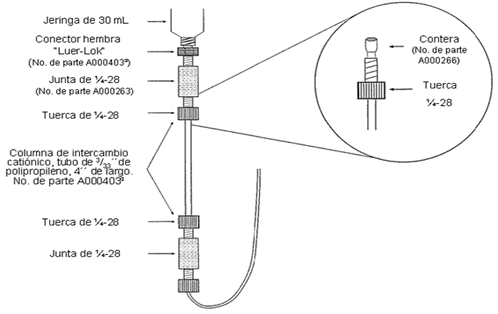 Figura B.11.2-3. Partes de la columna de intercambio catiónico. Las referencias a partes específicas de marcas o números de catálogo se incluyen sólo como ejemplos y no implican aprobación de los productos. Tomar con una jeringa de plástico de 20 mL o de mayor capacidad la resina de intercambio en suspensión y una vez llena, conectar la jeringa a la columna con ayuda del conector hembra "Luer-Lok". Colocar la jeringa en posición horizontal para que la resina no quede en la salida de ésta. Con cuidado presionar el émbolo de manera que la columna se llene de agua. Una vez que haya desplazado el aire del interior de la columna colocar la jeringa en posición vertical con lo que la resina comenzará a introducirse en la jeringa por gravedad. Si la resina deja de entrar en la columna agitar suavemente la jeringa o dar pequeños golpes en la base de la jeringa. Continuar hasta que la resina llene completamente la columna. Antes de retirar la jeringa, asegurarse de mantener el extremo opuesto de la manguera conectada a la columna en una posición más alta que el extremo de la columna por donde introdujo la resina. Esto con el fin de que cuando retire la jeringa, el agua fluya desde el interior de la columna y evite la entrada de aire. En caso de que quede aire atrapado en el interior de la columna, vaciarla y comenzar el procedimiento nuevamente. Retirar la jeringa junto con el conector hembra "Luer-Lok". Limpie los restos de resina que hayan quedado fuera de la columna de polipropileno. Con cuidado colocar el tapón poroso frit dentro de la contera y reestablezca las conexiones de la tuerca con la junta. Instalar de la columna de intercambio catiónico. Para evitar la entrada de aire en la columna de intercambio catiónico, instálela con la bomba en funcionamiento. Una vez que haya obtenido una línea base estable con todos los reactivos fluyendo por el sistema y con la bomba en funcionamiento, desconectar la tuerca que se encuentra entre los desburbujeadores conectados a los puertos 2 y 10 del cartucho dejando la junta conectada a la tuerca que viene del puerto 2 (o sus equivalentes). Retirar la junta de la entrada de la columna de intercambio ya preparada (el extremo que permaneció cerrado con el tapón poroso al momento de empacarla). Introducir y asegurar esta tuerca de la columna a la junta que viene del puerto 2. Retirar la tuerca en el otro extremo de la columna de intercambio dejando la junta conectada a ésta. Conectar aquí la tuerca en la manguera que va al puerto 10. Dejar que se estabilice el sistema (lo cual requiere de aproximadamente 20 minutos) y verifique que no entre aire en la columna de intercambio catiónico. Verifique nuevamente las condiciones de la línea base. Las referencias a partes específicas de marcas o números de catálogo se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden representar especificaciones adecuadas para los artículos. B.11.2.7.9 Procedimiento analítico Colocar la(s) gradilla(s) con los estándares y las muestras en el automuestreador. Verificar que las posiciones de los tubos coincidan con las especificadas en la secuencia de análisis. Los calibrantes, blancos y muestras sintéticas deben tener la misma matriz que las muestras analizadas. Iniciar el análisis presionando el botón que generalmente tiene el símbolo 8 (o su equivalente) ubicado en el margen izquierdo de la pantalla de obtención de datos. Esto inicia el análisis de acuerdo a la secuencia cargada. De ser necesario, realizar las diluciones de las muestras que se encuentren fuera del intervalo de trabajo e inclúyalas al final de la secuencia, puede apoyarse de la prueba presuntiva de sulfato para determinar la posible dilución previa. Anexar también al final de la secuencia las muestras para las que se hayan obtenido resultados poco confiables debido a anomalías durante su análisis, tal como el arrastre de la muestra anterior o el paso de alguna burbuja por el detector. Para anexar muestras a la secuencia abra la tabla durante el análisis del lote presionando el botón correspondiente ubicado en el margen izquierdo de la pantalla de obtención de datos e introduzca la información en las posiciones disponibles al final de la secuencia. Para realizar las diluciones utilice material verificado y/o calibrado, en caso que se presuma una concentración elevada de analito no debe hacer una dilución directa mayor a 20, ya que puede estar tomando una concentración de analito poco representativa y cometer un error en la dilución. En caso de hacer diluciones mayores a 20, se hace una serie de diluciones tomando como base la inicial dilución 20 que ya se realizó. No haga diluciones volumétricamente con material sin verificar. Una vez terminado el análisis, desmontar la columna de intercambio catiónico y reestablecer las conexiones de las tuercas con las juntas entre los puertos 2 y 10. Hacer pasar durante 15 minutos agua desionizada por la línea de NaOH y del diluyente, y disolución de lavado por la línea del reactivo de AMT de la misma manera que se hizo al arrancar el equipo. Detener la bomba, libere las grapas de las mangueras en la bomba peristáltica, apagar el equipo y vaciar el recipiente de desechos. B.11.2.7.10 Consideraciones operativas al procedimiento analítico La determinación de sulfato con el método de azul de metiltimol es inherentemente ruidosa. Para reducir el ruido en la línea base se recomienda: · Usar agua desionizada y/o desgasificada para la preparación de reactivos y para el enjuague del automuestreador. · Usar una sal de azul de metiltimol de calidad. De ser posible, solicitar al proveedor un certificado de este reactivo. Usar sólo azul de metiltimol con un contenido de agua menor al 10%. La cantidad de cationes interferentes varía con la matriz de cada muestra. Las muestras con una concentración alta de cationes multivalentes pueden requerir un reemplazo más frecuente de la resina de intercambio catiónico o una columna de intercambio catiónico más larga. La columna de intercambio catiónico se prepara cada vez que se realiza un lote, en caso que se reutilice se debe verificar la capacidad de intercambio de la columna analizando una disolución estándar de una concentración de calcio (Ca2+) similar a la de las muestras analizadas. Un recobro bajo indica que se ha sobrepasado la capacidad de la columna de intercambio catiónico y debe re-prepararse. La columna tiene una capacidad de intercambio de aproximadamente 35 mg de calcio, lo que representa alrededor de 900 muestras con una concentración de calcio de 200mg/L. La resina de la columna debe ser reemplazada tan frecuentemente como sea necesario, como para asegurar que no se use más del 50% de su capacidad. Después de haber analizado un gran número de muestras con concentraciones elevadas de sulfato, se puede observar un aumento en la deriva de la línea base. En ese caso es necesario limpiar la celda de flujo con disolución limpiadora y después enjuagarla con agua desionizada en abundancia. Si la línea base no mejora, limpiar todo el cartucho con disolución limpiadora. El reactivo de azul de metiltimol debe prepararse al menos 24 horas antes de usarse y permanecer almacenado en un frasco de vidrio ámbar a 4 °C. El pH del reactivo de azul de metiltimol de trabajo se debe ajustar entre 2.6 y 2.7 unidades de pH antes de usarse. El pH del medio al abandonar la celda de flujo debe ser de 12.5 unidades de pH. Cuando se instale la columna de intercambio catiónico, mantener en todo momento la bomba funcionando. Esto evita la entrada de aire en la columna. Si observa una línea base inestable, reemplazar la manguera de silicón del reactivo de azul de metiltimol. B.11.2.7.11 Análisis de datos y cálculos La curva de calibración permite al equipo calcular automáticamente la concentración de las muestras. Las unidades de los valores arrojados son las mismas que se usaron para los estándares, que son mg/L de SO42- B.11.2.7.12 Informe de prueba La expresión de resultados debe ser en  con dos cifras decimales. con dos cifras decimales. B.11.2.8 Control de calidad, criterios de aceptación y rechazo, calibración e interferencias B.11.2.8.1 Control de calidad B.11.2.8.1.1 Verificación del equipo. Para asegurar el buen funcionamiento del equipo revise los puntos indicados en el formato de Verificación de Indicadores de Calidad de la técnica. B.11.2.8.1.2 Verificación inicial de la calibración. Si para cuantificar la concentración de analito en las muestras y los controles de calidad se emplea una curva de calibración cargada en el programa del equipo obtenida previamente (durante la validación parcial del método), se emplea la respuesta que arroja el pico de sincronía para evaluar la veracidad de la curva. Se compara la respuesta obtenida para este pico con el promedio de las respuestas obtenidas en la validación del método para un estándar de la misma concentración. La diferencia entre estas dos repuestas no debe ser mayor al 15%. Si se hace uso de una curva de calibración nueva para cuantificar la concentración de analito en las muestras y los controles de calidad en un lote, se debe preparar un estándar de verificación inicial de la calibración (ICV), el cual contiene al analito en una concentración conocida y que se encuentra dentro del intervalo de trabajo. Ya que la curva de calibración para sulfato se ajusta a una ecuación de tercer orden, se recomienda que se empleen dos disoluciones ICV. La concentración de una de ellas preparada en la porción de concentraciones bajas (entre 10-100 y mg/L), y la segunda por arriba de 100 mg/L. Estos estándares deben ser preparados a partir de una fuente alterna a la que se empleó para construir la curva de calibración. B.11.2.8.1.3 Verificación de contaminación de reactivos. Muestras de agua: Se prepara un blanco de reactivos tomando una alícuota de agua desionizada y sometiéndola al mismo procedimiento de preparación y de análisis que el resto de las muestras. La lectura para este blanco de reactivos debe arrojar un valor menor al límite de detección del método y se analiza una muestra blanco por cada 20 muestras reales. B.11.2.8.1.4 Verificación del proceso analítico. Se emplean diferentes muestras para verificar el proceso analítico: · Verificación inicial de la calibración. · Verificación de calibración continua (CCC): Con el propósito de comprobar que la calibración verificada al inicio se mantenga vigente durante todo el análisis del lote, se debe analizar a intervalos regulares una disolución estándar de concentración conocida. Se sugiere que ésta sea de la misma concentración del punto intermedio de la curva. La diferencia entre el valor nominal de esta disolución y el valor obtenido en cada medición no debe ser mayor al 10%. · Verificación de la linealidad de la curva de calibración. El programa del equipo ajusta una ecuación a los pares de datos obtenidos para las muestras con las que se construyó la curva de calibración. Este ajuste es de tercer orden en el caso de sulfato. Además de la función ajustada, el programa arroja un coeficiente de correlación lineal, el cual debe ser igual o mayor a 0.995. · Se analiza una muestra sintética de concentración conocida, preparada por el analista preferentemente de una fuente diferente a la de la curva de calibración. La diferencia entre el resultado obtenido y el valor nominal de esta muestra no debe ser mayor al 20%. Se prepara una muestra sintética de concentración conocida por cada 20 muestras reales, estos estándares internos o muestras sintéticas de concentración conocida corresponden a muestras QC o blancos fortificados. · Verificación de repetibilidad de los análisis. Se selecciona al menos una muestra por cada matriz del lote analizado para prepararla y analizarla por duplicado. Con ello se verifica la repetibilidad del proceso al cual se someten las muestras. La diferencia porcentual relativa entre los dos valores obtenidos no debe superar el 20%. · Verificación de interferencias de matriz. Se debe adicionar al menos una muestra por cada matriz del lote analizado. Las muestras seleccionadas se adicionan con una concentración conocida de analito, la cual se recomienda que sea igual a la establecida como máximo permisible por alguna entidad reguladora o, en su caso, que sea entre una y cinco veces la concentración del analito presente originalmente en la muestra o bien una concentración aproximada del punto intermedio del intervalo de trabajo o menor. Comparando las concentraciones de analito en la muestra sin adicionar y en la adicionada se obtiene un recobro de la adición, el cual debe de estar entre el 80 y el 120 %. · Composición del lote. El lote analítico debe conformarse como se indica en la Tabla B.11.2-2, de este Apéndice.
B.11.2.8.1.5 A partir de que comienza el análisis de las muestras desconocidas, se debe incluir cada 10 muestras el siguiente grupo de inyecciones a lo largo de toda la secuencia para la verificación continua de la calibración: · Enjuague (BLNK) · Estándar de verificación de calibración continua (CCC) · Enjuague (BLNK) · Línea base (RB) Este grupo también se debe anexar al final de la secuencia inmediatamente después de la última muestra problema. Dependiendo de los requerimientos del programa específico de control de calidad, pueden ser requeridas muestras duplicadas de campo para evaluar la precisión y exactitud del muestreo, así como las técnicas de transportación y almacenamiento de la muestra. B.11.2.8.2 Criterios de aceptación y rechazo. La Tabla B.11.2-3, de este Apéndice resume los controles de calidad mencionados en esta sección, así como sus criterios de aceptación.
B.11.2.8.3 Calibración Si para cuantificar la concentración de SO42- se emplea una curva obtenida anteriormente en la validación parcial del método, solamente verifique la calibración del instrumento. Si se requiere de una curva de calibración nueva para cuantificar la concentración de analito en las muestras y en los controles de calidad de un lote, o para realizar una nueva validación parcial del método, prepárela a partir de la disolución madre usando material volumétrico verificado, o bien, gravimétricamente empleando una balanza calibrada. En la Tabla B.11.2-4, de este Apéndice se muestran las cantidades sugeridas para obtener una curva de calibración de SO42- en el intervalo de 5 a 200 mg/L a partir de la disolución madre de Na2SO4. Preparar una disolución intermedia de 1 000 mg/L de sulfato, partiendo de la disolución madre de 10 000 mg/L. Se toman 10mL de disolución madre y se aforan a 100 mL con agua desionizada. Este procedimiento puede hacerse volumétrica o gravimétricamente únicamente garantizando la verificación y/o calibración de los instrumentos y equipo utilizados.
Preparar el equipo para el análisis de muestras e incluir en la secuencia los puntos de calibración preparados, introduciendo las concentraciones calculadas para cada punto en la ventana referente a las condiciones del método. Si va a utilizar una curva de calibración cargada en el equipo para la cuantificación debe preparar una disolución de concentración igual al del último punto de la curva para comparar la respuesta obtenida contra la que se está cuantificando (Preferentemente la de la confirmación del método). En este caso también se debe preparar una disolución para la verificación de la calibración continua, la cual puede ser de una concentración de magnitud igual o menor al punto medio de la curva, idealmente se deben preparar dos estándares de diferente concentración (uno en la alta y uno en la baja del IT) para asegurar que la calibración sigue vigente en todo el IT a lo largo de la corrida. En estos casos no se requiere de la determinación del ICV. El equipo registra la respuesta obtenida para cada punto y la asocia a la concentración especificada por el usuario, con lo que construye y almacena de manera automática una curva de calibración. Esta curva es usada después para obtener la concentración de las muestras desconocidas. El ajuste para la curva de calibración de sulfato no es lineal, sino de tercer orden. La verificación de la calibración durante el análisis del lote se realiza mediante la CCC. B.11.2.8.4 Interferencias Al tratarse de una técnica fotométrica, la turbidez en las muestras interfiere con la capacidad del detector para registrar la absorción real de la muestra, por lo cual las muestras turbias deben filtrarse antes de analizarlas. Los cationes multivalentes en general, y particularmente los que se asemejan al bario (Ba2+) tales como el calcio (Ca2+) y el magnesio (Mg2+) generan una interferencia, por lo que pueden ser eliminados de la muestra al pasarla por la columna de intercambio catiónico. Si el pH de la muestra es muy bajo, la concentración tan alta de iones H+ puede ocasionar la liberación de los cationes retenidos en la columna de intercambio, por lo que es necesario neutralizar las muestras cuyo pH sea menor a 2 unidades de pH. B.11.2.9 Seguridad y manejo de residuos B.11.2.9.1 Seguridad Cada reactivo utilizado en estos procedimientos debe ser tratado como un riesgo potencial para la salud y la exposición a estos materiales debe ser minimizada. Cada laboratorio es responsable de mantener un conocimiento de las regulaciones respecto a la manipulación segura de cualquier producto químico usado en este método. Debe ponerse a disposición de todo el personal involucrado en el análisis las hojas de datos de seguridad de los productos químicos. Debe de utilizarse equipo de protección personal apropiado para el manejo de muestras y estándares. Los estándares primarios deberán prepararse en una campana. Cuando el analista maneje altas concentraciones de compuestos tóxicos deberá utilizarse un respirador de gases tóxicos. La carcinogenicidad de todos los reactivos no ha sido determinada con precisión, por lo que cada sustancia debe tratarse como potencialmente peligrosa para la salud. La exposición a estas sustancias debe reducirse al menor nivel posible. Las muestras desconocidas pueden ser peligrosas. Manéjelas siempre con precaución extrema. Cuando se trabaje con muestras desconocidas o con los reactivos empleados en este método use siempre el equipo de seguridad adecuado como bata de trabajo, lentes de seguridad y guantes apropiados. B.11.2.9.2 Manejo de residuos Es la responsabilidad del laboratorio cumplir con todos los reglamentos federales, estatales y locales referentes al manejo de residuos, particularmente las reglas de identificación, almacenamiento y disposición de residuos peligrosos. B.11.2.10 Referencias Dirección General de Normas. NMX-AA-074-1981. Análisis de agua Determinación del Ion Sulfato. Dirección General de Normas. NMX-B-021-1982. Determinación de las formas de azufre en el carbón. O.I. Analytical. 2008. Sulfate by Segmented Flow Analysis (SFA). U.S. Environmental Protection Agency. 1986. EPA Method 9036. Sulfate (Colorimetric, Automated, Methyltimol Blue, AA II). B.12 MÉTODO DE PRUEBA PARA LA DETERMINACIÓN DE SÓLIDOS DISUELTOS TOTALES (SDT) EN AGUA PARA USO Y CONSUMO HUMANO B.12.1 Principio Una muestra bien mezclada se pasa a través de un filtro de fibra de vidrio estándar y el filtrado se coloca en un recipiente llevado a peso constante mediante secado a 180 °C. El incremento en el peso del plato representa el total de sólidos disueltos (SDT). Este procedimiento puede utilizarse para el secado a otras temperaturas (103 a 105 °C). B.12.2 Alcance y aplicación Este método es utilizado para la determinación de sólidos disueltos totales secados a 180°C en agua para uso y consumo humano. B.12.3 Equipos y materiales Sólo se mencionan los equipos y materiales que no son de uso común en el laboratorio analítico. Todo el material volumétrico utilizado debe ser clase A o estar verificada su calibración Agitador magnético con barra de agitación TFE. Balanza analítica Baño de vapor Desecador, provisto de un desecante que contiene un indicador de color para la concentración de humedad o un indicador instrumental. Discos filtrantes de fibra de vidrio sin aglutinante orgánico. Dispositivo de filtración, adecuado para el disco de filtro seleccionado, pudiendo ser embudo de filtro de membrana; crisol Gooch de 25 a 40ml de capacidad, con el adaptador de crisol Gooch; o aparato de filtración con depósito y disco fritado grueso (40-60 mm) como filtro de apoyo. Recipiente receptor del filtrado aplicando vacío, de capacidad adecuada para el volumen total de muestra seleccionado y enjuages. Horno de secado, para funcionamiento a 180±2°C (o 103 a 105 °C). Pipetas volumétricas Platos de evaporación, platos o cápsulas de 100mL de capacidad de porcelana, de níquel, de platino o de vidrio alto en sílice, de 90 mm de diámetro. B.12.4 Procedimiento B.12.4.1 Preparación del disco de filtro de fibra de vidrio Insertar el disco en el aparato de filtración. Aplicar vacío y lavar con tres volúmenes sucesivos de agua reactivo de 20mL, permitir el drenaje completo entre lavados y continuar la succión durante 3 minutos más. B.12.4.2 Preparación del plato o cápsula de evaporación Secar el plato o cápsula a 180±2°C (a 103 a 105 °C) durante 1 hora en una estufa. Enfriar en el desecador el tiempo necesario. Pesar inmediatamente antes de usar. B.12.4.3 Selección de los tamaños del filtro y de la muestra Elegir el volumen de la muestra para obtener entre 2.5 y 200 mg de residuo seco, generalmente se cumple con 100 mL de muestra. Si se requieren más de 10 minutos para completar la filtración, aumentar el tamaño del filtro o disminuir el volumen de la muestra. B.12.4.4 Procedimiento analítico Agitar la muestra con un agitador magnético y pipetear un volumen medido sobre un filtro de fibra de vidrio previamente lavado, con vacío aplicado hacia un recipiente limpio de tamaño adecuado al volumen de las muestras y los lavados. Lavar con tres volúmenes sucesivos de 10ml de agua de grado reactivo, lo que permite el drenaje completo entre lavados, y continuar la succión durante unos 3 minutos después de que la filtración se ha completado. Transferir el filtrado total con lavados a un plato o cápsula de evaporación previamente pesado y preparado como se indicó anteriormente, evaporar a sequedad en un baño de vapor o en una estufa de secado. Cuando el tamaño de la porción de la muestra es mayor que la capacidad de la l plato o cápsula de evaporación, agregaras las porciones de muestra sucesivas al mismo plato después de secar el filtrado anterior. Secar la muestra evaporada durante al menos 1 hora en una estufa a 180±2 °C (o 103 a 105 °C), enfriar en un desecador y pesar. Repetir el ciclo de secado, enfriamiento, desecación y pesaje hasta que se obtenga un peso constante con una diferencia de 2 mg. Analizar muestras por duplicado. Las determinaciones duplicadas deben coincidir en un ±5% de su peso promedio. B.12.4.5 Análisis de datos y cálculos Calcular la concentración de sólidos disueltos totales utilizando la siguiente fórmula: 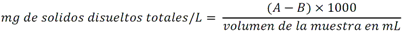 En donde: A peso del residuo seco + peso de plato o cápsula vacía, mg B peso de plato o cápsula vacía, mg B.12.5 Informe de prueba La expresión de resultados debe ser en  con dos cifras decimales. con dos cifras decimales. B.12.6 Referencias Standard methods for examination of water and wastewater. 2540 C Total Dissolved Solids Dried at 180°C. B.13 MÉTODO DE PRUEBA PARA LA DETERMINACIÓN DE COMPUESTOS ORGÁNICOS NO HALOGENADOS EN AGUA PARA USO Y CONSUMO HUMANO B.13.1 Símbolos y términos abreviados CG/FIC cromatografía de gases con detector de ionización de flama CONH compuestos orgánicos no halogenados FC factor de calibración H2SO4 ácido sulfúrico NaOH hidróxido de sodio B.13.2 Principio Este método contiene los procedimientos de extracción, condiciones cromatográficas, ventanas de tiempo, requerimientos de control de calidad y cálculos para la identificación y cuantificación de compuestos orgánicos no halogenados. Los compuestos orgánicos no halogenados en matriz agua, son extraídos con cloruro de metileno por extracción líquido-líquido, el extracto es analizado por cromatografía de gases inyectando de 1 a 3 µL. B.13.3 Alcance y aplicación Este método es utilizado para la determinación de compuestos orgánicos no halogenados, que comprende el intervalo de punto de ebullición de compuestos orgánicos con esturcturas de 10 carbonos (C10) a 28 carbonos (C28), extraíbles por cromatografía de gases con detector de ionización de flama (CG/FID) en muestras de agua para uso y consumo humano. B.13.4 Equipos y materiales Las referencias a marcas específicas y números de catálogo se incluyen sólo como ejemplos y no implican aprobación de los productos. Dicha referencia no excluye el uso de otros proveedores o fabricantes. Las referencias específicas pretenden representar especificaciones adecuadas para los artículos. Sólo se mencionan los equipos y materiales que no son de uso común en el laboratorio analítico. Todo el material volumétrico utilizado debe ser clase A o estar verificada su calibración. Baño María con control de temperatura. Caja de papel filtro Whatman no. 41 Cromatógrafo de gases con detector de ionización de flama (CG/FID), equipado con puerto de inyección capilar y automuestreador, así como columna capilar de 30 m, 25 mm de diámetro y película de metil silicón de 1.0 µm de espesor. Embudos de filtración de tallo corto o largo. Equipos concentradores kuderna-danish (tubo concentrador, matraz de 500 mL y columna snyder de tres bolas). Gradilla para viales de 2 mL. Matraz Erlenmeyer de 250 mL. Matraz de separación con llave de teflón de 2 litros. Matraz volumétrico de 1, 5 y 10 mL. Microjeringas de 10, 25, 50, 100, 500 y 1 000 µL Parrilla de agitación magnética Parrilla de calentamiento Perlas de ebullición Pipetas Pasteur de 230 mm. Probetas graduadas de 10 y 100 mL y de 1 L. Termoblock Vasos de precipitados de 100, 250 mL y 500 mL. Viales de 2 mL aforados a 1 mL, transparentes. Vortex. B.13.5 Reactivos y soluciones Los reactivos que requiere el método deben ser tipo ACS grado reactivo a menos que otra cosa se indique. Cloruro de metileno grado plaguicida. B.13.6 Procedimiento B.13.6.1 Preparación de disoluciones de calibración La preparación de las disoluciones de calibración puede realizarse de forma gravimétrica. Estándar de Compuestos Orgánicos No Halogenados (CONH) de 20 000 mg/L. Mezcla de hidrocarburos lineales que contenga los siguientes hidrocarburos con estructuras de 10, 12, 14, 16, 18, 20, 22, 24, 26 y 28 carbones respectivamente (C10, C12, C14, C16, C18, C20, C22, C24, C26 y C28) con una concentración de 2 000 mg/L de cada compuesto (20 000 mg/L de concentración total). Estándar de CONH de 1 000 mg/L. Tomar 250 µL de la solución de CONH de 20 000 mg/L en un matraz volumétrico de 5 mL y aforar con cloruro de metileno. Estándar de CONH de 100 mg/L. Tomar 100 µL de la solución estándar de CONH de 1 000 mg/L en un matraz volumétrico de 1 mL y aforar con cloruro de metileno. Estándar de CONH de 10 mg/L. Tomar 100 µL de la solución estándar de CONH de 100 mg/L en un matraz volumétrico de 1 mL y aforar con cloruro de metileno. Estándar de CONH de 1 mg/L. Tomar 100 µL de la solución estándar de CONH de 10 mg/L en un matraz volumétrico de 1 mL y aforar con cloruro de metileno. Estándar de CONH de 0.1 mg/L. Tomar 100 µL de la solución estándar de CONH de 1 mg/L en un matraz volumétrico de 1 mL y aforar con cloruro de metileno (QUINTO PUNTO DE CURVA). Estándar de CONH de 0.05 mg/L. Tomar 50 µL de la solución Estándar de CONH de 0.1 mg/L en un matraz volumétrico de 1 mL y aforar con cloruro de metileno (CUARTO PUNTO DE CURVA). Estándar de CONH de 0.02 mg/L. Tomar 200 µL de la solución estándar de CONH de 0.1 mg/L en un matraz volumétrico de 1 mL y aforar con cloruro de metileno (TERCER PUNTO DE CURVA). Estándar de CONH de 0.01 mg/L. Tomar 100 µL de la solución estándar de CONH de 0.1 mg/L en un matraz volumétrico de 1 mL y aforar con cloruro de metileno (SEGUNDO PUNTO DE CURVA). Estándar de CONH de 0.005 mg/L. Tomar 500 µL de la solución estándar de CONH de 0.01 mg/L en un matraz volumétrico de 1 mL y aforar con cloruro de metileno (PRIMER PUNTO DE CURVA). B.13.6.2 Preservación y extracción de las muestras Enjuagar el material de vidrio con cloruro de Metileno, Sonicar los magnetos con cloruro de metileno durante dos minutos, repetir el proceso tres veces. Tomar 1L de cada muestra en matraz de separación de 2 L, incluyendo blanco de reactivos, muestras control, muestra adicionada y muestras reales. Medir el pH de cada muestra y ajustar si es necesario con ácido sulfúrico (H2SO4) 1:1 o con hidróxido de sodio (NaOH) 10 N de modo que el pH quede en el intervalo de 5 a 9. Agregar 50 mL de cloruro de metileno, agitar durante tres minutos, después de la agitación dejar reposar para permitir la separación de las fases, recuperar el cloruro metileno en un matraz Erlenmeyer de 250 mL, pasándolo previamente por un embudo de filtración con fibra de vidrio o papel filtro No. 41 y sulfato de sodio anhidro purificado. Si la muestra presentara problemas de emulsión durante la extracción se deberán utilizar procedimientos mecánicos de separación, como centrifugación, filtración, etc. Extraer dos veces más con 25 mL de cloruro de metileno como en el inciso anterior y los extractos se recuperan en el mismo matraz Erlenmeyer. Concentrar los extractos en Kuderna-danish en baño María a una temperatura entre 60 y 65°C y hasta que el volumen aparente del extracto sea menor de 1 mL (no dejar secar el extracto ya que se pierden los compuestos orgánicos semivolátiles ligeros), posteriormente retirar la columna Snyder y con una pipeta pasteur recuperar el extracto en vial aforado de 2 mL, aforar a la marca de 1 mL con evaporación con nitrógeno a temperatura ambiente y proceder al análisis cromatográfico. B.13.6.3 Análisis cromatográfico Las condiciones para el análisis cromatográfico se muestran en la Tabla B.13-1, de este Apéndice, para alcanzar la rampa de temperatura de 75 °C/minutos se cuenta con un GC Racer, sin embargo, las condiciones cromatográficas dependen de la columna y el equipo utilizado, por lo que pueden variar para optimizar la resolución y tiempo de análisis.
B.13.6.4 Análisis de datos y cálculos La concentración de la muestra (C) se obtiene de extrapolar el área total de los picos en la ventana cuantitativa utilizando la siguiente ecuación: 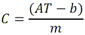 En donde: C concentración en mg/L AT área total en la ventana cuantitativa b ordenada al origen de la regresión lineal de la curva de calibración m pendiente de la regresión lineal de la curva de calibración Una vez calculada la ecuación anterior, la concentración para muestras de aguas se determinará en mg/L con la siguiente ecuación: 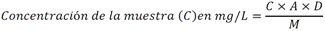 En donde: C concentración de la muestra en mg/L A volumen de aforo o volumen de concentración final del extracto en L D factor de dilución del extracto M volumen de la muestra extraída en L B.13.6.5 Informe de prueba Reportar los compuestos orgánicos no halogenados (CONH) en mg/L con tres cifras decimales, el cual es la suma de los compuestos hidrocarburos C10, C12, C14, C16, C18, C20, C22, C24, C26 y C28. B.13.7 Control de calidad, especificaciones de aceptación y rechazo, calibración e interferencias B.13.7.1 Control de calidad B.13.7.1.1 Verificación del equipo. Antes del análisis de cada lote analítico verificar que el instrumento esté libre de interferencias ocasionadas por inyecciones previas de muestras muy concentradas y sangrado de columna, contaminación por gases o por jeringa sucia, para tal efecto se realiza un blanco electrónico que consiste en hacer una corrida del método a utilizar, pero sin inyectar nada. El blanco electrónico no deberá presentar ningún pico ni elevación o bajas de línea base y en caso contrario se procederá a purgar el sistema con temperatura del horno a 300 °C durante 15min. B.13.7.1.2 Verificación de la calibración inicial. La curva de calibración inicial deberá realizarse inyectando los estándares de calibración preparados y se considera lineal, si el por ciento de la desviación estándar relativa (%DER) de los factores de calibración (FC) es menor o igual al 15%, el cálculo de FC se realiza con la siguiente ecuación: 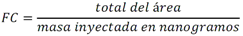 La verificación de la calibración inicial puede también efectuarse mediante la regresión de mínimos cuadrados, en la cual el criterio de aceptación es que el coeficiente de correlación (r) sea mayor o igual a 0.99. Si los criterios para % de RSD y r no se cumplen se procede a la revisión del sistema cromatográfico o a la preparación de las disoluciones de calibración. Posteriormente la curva de calibración inicial, será aceptada si el análisis de un punto intermedio de la curva no presenta una variación de la concentración mayor del 15% y que será calculada de la siguiente manera: 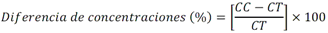 En donde: CC concentración calculada CT concentración teórica 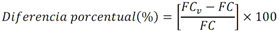 En donde: FCv factor de calibración respuesta (cualquiera que aplique) de los compuestos del estándar de verificación. FC media del factor de calibración o la media del factor de respuesta de la calibración Inicial. Si el criterio de aceptación para la diferencia de la concentración es mayor a 15% proceder a la revisión del sistema cromatográfico o a la preparación de la solución estándar utilizada (concentración media de la curva de calibración). B.13.7.1.3 Verificación de contaminación de reactivos. Para cada lote de análisis preparar un blanco de reactivos, procesando agua tipo I de la misma forma en que se procesan las muestras reales, utilizando los mismos solventes y reactivos. El blanco de reactivos no deberá presentar ningún pico a excepción del pico del solvente, en caso de que se presenten picos en el cromatograma proceder a la localización de la fuente de contaminación (solventes, reactivos o material). B.13.7.1.4 Verificación del proceso analítico. Verificar que la recuperación promedio de la muestra de control de calidad esté entre el 70% y el 130%, en caso de no cumplir con el criterio, proceder a la revisión del sistema cromatográfico, a la preparación de la muestra control, etc. Determinar el error, corregir y continuar con el análisis. B.13.7.1.5 Verificación de interferencia de matriz. Analizar muestras adicionadas. Verificar que la recuperación promedio de las muestras adicionadas esté entre 70 y 130 %, de no ser así, revisar el sistema cromatográfico, etc. Determinar la variación, corregir y continuar con el análisis. B.13.7.1.6 Verificación de la precisión del proceso analítico. La precisión del lote analítico se realiza mediante el análisis de una muestra por duplicado. Calcular la diferencia porcentual relativa entre la muestra y la muestra duplicada, el valor deberá ser menor al 20%: 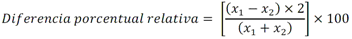 En donde: X1 valor de concentración del compuesto en la muestra. X2 valor de concentración del compuesto en la muestra duplicada. A.2.13.7.1.7 Verificación de la estabilidad de la curva de calibración. Analizar un estándar de concentración media de la curva y un blanco de reactivos, cada 20 muestras o 12 horas de análisis, y verificar el porcentaje de la variación de la concentración y proceder como en la verificación de la calibración inicial. B.13.7.2 Criterios de aceptación y rechazo B.13.7.2.1 Verificación del equipo. Equipo libre de interferencias (blanco electrónico). No debe presentar ningún pico o perfil de picos en ningún tiempo de retención. B.13.7.2.2 Verificación de la calibración inicial. Evaluación del estándar de verificación (punto intermedio de la curva de calibración; % de diferencia de la concentración. Menor al 15%. B.13.7.2.3 Verificación del proceso analítico. Blanco de Reactivos. No debe presentar ningún pico o perfil de picos al tiempo de retención del analito a identificar. Evaluación de muestra de control de calidad (MCC). La recuperación de la concentración de la muestra control se deberá encontrar dentro del criterio de aceptación de 70-130 %. B.13.7.2.4 Verificación de interferencia de matriz. Evaluación de muestras adicionadas. % de recuperación del 70 130%. B.13.7.2.5 Verificación de la precisión del método. Evaluación de muestras duplicadas. La recuperación de la muestra duplicada deberá cumplir con el criterio de aceptación 20%. B.13.7.3 Calibración. B.13.7.3.1 Estabilidad de la curva de calibración. Evaluación del estándar de verificación (punto intermedio de la curva de calibración; % de diferencia de la concentración, menor al 15%. B.13.7.3.2 Calibración Inicial. El procedimiento utilizado para la calibración de este método es el de estándar externo y se inyectarán de 1 a 3 µL por triplicado de cada uno de los niveles requeridos, al final realizar el promedio de las tres curvas y agregarlo de forma manual a la tabla de calibración del método. B.13.7.3.3 Preparación de la curva de calibración. El volumen a usar de las disoluciones a partir de la cual se preparan los puntos de la curva puede ser calculado con la siguiente ecuación: 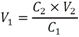 En donde: V1 volumen en mL a partir de la disolución patrón. C1 concentración total requerida para cada punto de la curva de calibración en mg/L. C2 concentración de la disolución de referencia que es la suma de las concentraciones de todos los compuestos de la disolución (C10 a C28) en mg/L. V2 volumen final (aforo) en mL de cada punto de la curva de calibración. A partir de la disolución patrón, calcular la masa que será utilizada para preparar los diferentes niveles de la curva de calibración en una masa total de 1 g, de acuerdo a la siguiente ecuación. 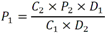 En donde: P1 peso de la disolución de referencia (g). C1 concentración de la solución de referencia que es la suma de las concentraciones de todos los compuestos de la disolución (C10 a C28) en mg/L. C2 concentración total del nivel requerido, que es la suma de las concentraciones de todos los compuestos de la disolución (C10 a C28) en mg/L. P2 peso de aforo del nivel requerido (g), es la suma de la masa de la disolución patrón y la masa del disolvente (g). D1 densidad de la disolución de patrón (g/mL). D2 densidad del disolvente de aforo (g/mL). A continuación calcular la concentración real de cada uno de los niveles de concentración utilizando la siguiente ecuación: 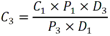 En donde: C3 concentración real de cada nivel (mg/L). C1 concentración de la solución de referencia que es la suma de las concentraciones de todos los compuestos de la disolución (C10 a C28) en mg/L. P1 peso de la mezcla de referencia utilizada (g). P3 peso de aforo (estándares + disolvente) en g. D1 densidad de la mezcla de referencia (g/mL). D3 densidad del disolvente de aforo (g/mL). B.13.7.3.4 Linealidad de la calibración. La calibración será lineal, si el porcentaje de la desviación estándar (%DER) de los factores de calibración es menor que el 20% sobre el rango de trabajo, la linealidad a través del origen puede asumirse y el promedio del factor de calibración puede usarse en lugar de una curva de calibración. Calcular el factor de correlación (r), si se utilizó una regresión de mínimos cuadrados, el cual deberá de tener un valor > 0.99. Si el valor de %DER es mayor al 20 % o el valor de r es menor a 0.99, la linealidad no puede asumirse y se procederá a encontrar las causas, corregirlas y reanalizar para cada nivel de calibración. B.13.7.4 Interferencias Cuando se tienen muestras con hidrocarburos pesados es muy posible que éstos se arrastren y contaminen inyecciones subsecuentes, provocando la aparición de picos o elevaciones de la línea base que no corresponden a las muestras que se analizan posteriormente. Para evitar este tipo de interferencias se recomienda purgar el equipo a 300 °C durante 15 minutos, después del análisis de una muestra muy concentrada, o hacer corridas de cloruro de metileno antes de inyectar otras muestras. B.13.8 Referencias EPA. 2007. Method 8015C (SW-846): Nonhalogenated Organics Using GC/FID. Rev. 3. APÉNDICE C INFORMATIVO Procesos propuestos para la potabilización del agua.
______________________________ |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||